| GDB-17 | |
|---|---|
| Basic Information | |
| Full Name | Generated Database 17 |
| Domain | Computational Chemistry |
| Year | 2012 |
| Publication & Access | |
| Paper | DOI |
| Dataset | gdb.unibe.ch |
| Dataset Composition | |
| Total Size | 166,443,860,262 molecules |
| CNO Molecules | 110.4 billion molecules |
| Full Set (with S, Halogens) | 166.4 billion molecules |
| Leadlike Subset (GDBLL-17) | 29 billion molecules |
| Technical Details | |
| Format | SMILES strings |
| Research Context | |
| Authors | Lars Ruddigkeit, Ruud van Deursen, Lorenz C. Blum, Jean-Louis Reymond |
| Institution | University of Berne, Ecole Polytechnique Fédérale de Lausanne |
GDB Series Overview: The Generated Database (GDB) series represents a systematic exploration of chemical space by generating all possible molecular structures. GDB-11 (26M molecules) established the methodology, GDB-13 (977M molecules) achieved billion-scale generation, and GDB-17 (166B molecules) represents the current limit of systematic chemical space generation.
Dataset Summary
GDB-17 contains 166.4 billion organic small molecules created by systematically exploring all possible structures with up to 17 atoms of carbon, nitrogen, oxygen, sulfur, and halogens. Building on the methodology from GDB-11 and GDB-13, this represents the largest systematically generated chemical database of its time. The dataset explores a vast space of molecular structures with high diversity. All molecules are provided as SMILES strings.
Related Databases: GDB-17 is the largest database in the Generated Database (GDB) series, following GDB-11 (26 million molecules) and GDB-13 (977 million molecules), demonstrating how far systematic chemical space enumeration can be scaled.
Key Features
- Large Scale: 166.4 billion molecules, greatly expanding known chemical space
- Complete Coverage: Systematically covers all possible structures from first principles, avoiding bias from existing databases
- More Elements: Includes sulfur and halogens (F, Cl, Br, I) beyond carbon, nitrogen, oxygen
- 3D Diversity: Contains more spherical molecules compared to the flat structures common in existing databases
- High Novelty: Contains 35× more molecular frameworks and 61× more ring systems than equivalent molecules from PubChem
- Series Culmination: Represents the current limit of the GDB methodology
Dataset Structure
The database is primarily defined by its total size, but two important “leadlike” subsets were also characterized for drug discovery applications.
| Category | Composition | Count | Description |
|---|---|---|---|
| Total Molecules | C, N, O, S, Halogens | 166.4 B | The complete enumerated database of molecules up to 17 heavy atoms. |
| Leadlike (GDBLL-17) | C, N, O, S, Halogens | 29 B | Subset with properties typical for lead compounds (100<MW<350, 1<clogP<3). |
| Leadlike, No Small Rings (GDBLLnoSR-17) | C, N, O, S, Halogens | 22 B | The GDBLL-17 subset with strained 3- and 4-membered rings removed. |
Structural Diversity
- Topology: Rich in complex ring structures. Molecules with three or more rings account for 32% of GDB-17, far more than in known databases. Simple molecules with no rings are rare (1.8%).
- Compound Types: Dominated by non-aromatic heterocycles (57%). Aromatic molecules, which make up a third of known compounds, are very rare (0.8%).
- Stereochemistry: The prevalence of non-flat structures means GDB-17 molecules are highly chiral. They average 6.4 stereoisomers per molecule—three times more than molecules in PubChem. Over 44% have eight or more possible stereoisomers.
Example Sample
The following is an example of a novel, previously unknown C₁₇-hydrocarbon ring system found in GDB-17.
C1CC2C3CCCC3C3(C4CCC3CC4)C2C1
Visualized with PubChem Sketcher:
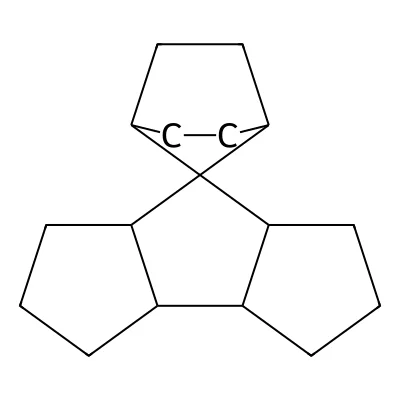
Representative GDB-17 molecule (SMILES: C1CC2C3CCCC3C3(C4CCC3CC4)C2C1) demonstrating the complex polycyclic structures and 3D diversity characteristic of the database
Use Cases
Primary Applications
- Virtual Screening: Shape-based screening and active site complementarity
- Scaffold Hopping: Discovering new molecular series from structural isomers
- Fragment-Based Discovery: Starting points for complex drug candidate building
Research Applications
- Chemical Space Exploration: Analyzing fundamental properties of small molecule universe
- AI/ML Benchmarking: Training and validating chemoinformatics tools and models
- Structure-Property Studies: 3D shape, stereochemistry, and scaffold complexity relationships
Quality & Limitations
Strengths
- Large Scale: Most complete systematic generation to date (166 billion molecules)
- High Novelty: Significantly more scaffolds and ring systems than existing databases
- 3D Diversity: Rich in spherical molecules, “escape from flatland”
- Series Achievement: Final development of GDB series methodology
- Coverage: Complete coverage of all possible structures with up to 17 atoms
Limitations
- Limited Atom Types: Excludes P, Si, B and other drug-relevant elements (expanded from GDB-11/GDB-13)
- Structural Constraints: Highly strained graphs, allenes, and nonaromatic C=C bonds excluded
- Functional Group Gaps: Wide range of unstable groups filtered out (hemiacetals, enols, aminals)
- Virtual Molecules: Computationally generated without experimental validation
- Synthetic Accessibility: No explicit scoring for synthetic feasibility
Generation and Filtering Pipeline
GDB-17 represents the current limit of the methodology established in GDB-11 and refined in GDB-13, achieving a thousand-fold increase over GDB-13 while maintaining chemical quality. The construction required a sophisticated, multi-step pipeline designed to ensure that the generated molecules are chemically stable and meaningful:
- Graph Generation & Selection: The process started with over 114 billion mathematical graphs (node-edge diagrams) with up to 17 nodes. These were filtered using topological and geometric criteria to remove highly strained and complex structures (e.g., fused small rings), resulting in 5.4 million stable hydrocarbon graphs.
- Skeleton Generation: Double and triple bonds were systematically added to the hydrocarbon graphs to create 1.3 billion unique unsaturated “skeletons”. Filters were applied to remove unstable arrangements like allenes and triple bonds in small rings.
- CNO Molecule Generation: The carbon atoms in the skeletons were combinatorially substituted with nitrogen and oxygen atoms, respecting valency rules. This generated over 110 billion molecules, which were then curated with a set of 12 chemical filters to remove unstable functional groups (e.g., enols, hemiacetals).
- Post-Processing & Diversification: Additional chemical diversity was introduced in a final step. Specific functional groups were systematically transformed to add sulfur, halogens, nitro groups, and oximes, adding another 56 billion molecules to yield the final database of 166.4 billion structures.
Citation: Ruddigkeit, L., van Deursen, R., Blum, L. C., & Reymond, J.-L. “Enumeration of 166 Billion Organic Small Molecules in the Chemical Universe Database GDB-17” J. Chem. Inf. Model. 2012, 52 (11), pp 2864–2875.