Paper Information
Citation: Qian, Y., Guo, J., Tu, Z., Li, Z., Coley, C. W., & Barzilay, R. (2023). MolScribe: Robust Molecular Structure Recognition with Image-To-Graph Generation. Journal of Chemical Information and Modeling, 63(7), 1925-1934. https://doi.org/10.1021/acs.jcim.2c01480
Publication: Journal of Chemical Information and Modeling 2023
Additional Resources:
Contribution: Generative Image-to-Graph Modelling
This is a Methodological Paper ($\Psi_{\text{Method}}$) with a secondary contribution to Resources ($\Psi_{\text{Resource}}$).
It proposes a novel architecture (image-to-graph generation) to solve the Optical Chemical Structure Recognition (OCSR) task, validating it through extensive ablation studies and comparisons against strong baselines like MolVec and DECIMER. It also contributes a new benchmark dataset of annotated images from ACS journals.
Motivation: Limitations in Existing OCSR Pipelines
Translating molecular images into machine-readable graphs (OCSR) is challenging due to the high variance in drawing styles, stereochemistry conventions, and abbreviated structures found in literature.
Existing solutions face structural bottlenecks:
- Rule-based systems (e.g., OSRA) rely on rigid heuristics that fail on diverse styles.
- Image-to-SMILES neural models treat the problem as captioning. They struggle with geometric reasoning (which is strictly required for chirality) and struggle to incorporate chemical constraints or verify correctness because they omit explicit atom locations.
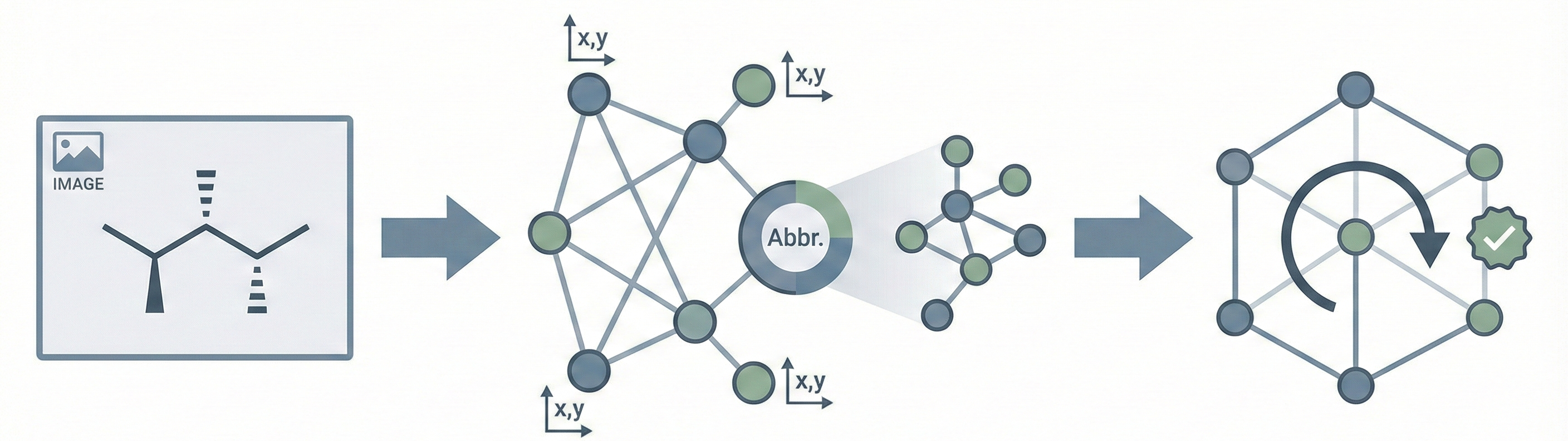
Core Innovation: Joint Graph and Coordinate Prediction
MolScribe introduces an Image-to-Graph generation paradigm that combines the flexibility of neural networks with the precision of symbolic constraints. It frames the task probabilistically as:
$$ P(G | I) = P(A | I) P(B | A, I) $$
Where the model predicts a sequence of atoms $A$ given an image $I$, followed by the bonds $B$ given both the atoms and the image.
- Explicit Graph Prediction: It predicts a sequence of atoms (with 2D coordinates) and then predicts bonds between them.
- Symbolic Constraints: It uses the predicted graph structure and coordinates to strictly determine chirality and cis/trans isomerism.
- Abbreviation Expansion: It employs a greedy algorithm to parse and expand “superatoms” (e.g., “CO2Et”) into their full atomic structure.
- Dynamic Augmentation: It introduces a data augmentation strategy that randomly substitutes functional groups with abbreviations and adds R-groups during training to improve generalization.
Methodology: Autoregressive Atoms and Pairwise Bonds
The authors evaluate MolScribe on synthetic and real-world datasets, focusing on Exact Match Accuracy of the canonical SMILES string. The model generates atom sequences autoregressively:
$$ P(A | I) = \prod_{i=1}^n P(a_i | A_{<i}, I) $$
To handle continuous spatial locations, atom coordinates map to discrete bins (e.g., $\hat{x}_i = \lfloor \frac{x_i}{W} \times n_{\text{bins}} \rfloor$), and decode alongside element labels. Bonds act on a pairwise classifier over the hidden states of every atom pair:
$$ P(B | A, I) = \prod_{i=1}^n \prod_{j=1}^n P(b_{i,j} | A, I) $$
- Baselines: Compared against rule-based (MolVec, OSRA) and neural (Img2Mol, DECIMER, SwinOCSR) systems.
- Benchmarks:
- Synthetic: Indigo (in-domain) and ChemDraw (out-of-domain).
- Realistic: Five public benchmarks (CLEF, JPO, UOB, USPTO, Staker).
- New Dataset: 331 images from ACS Publications (journal articles).
- Ablations: Tested performance without data augmentation, with continuous vs. discrete coordinates, and without non-atom tokens.
- Human Eval: Measured the time reduction for chemists using MolScribe to digitize molecules vs. drawing from scratch.
Results: Robust Exact Match Accuracy
- State-of-the-Art Performance: MolScribe achieved 76-93% accuracy across diverse benchmarks, significantly outperforming baselines (e.g., on the difficult Staker dataset, MolScribe achieved 71.9% compared to the next best 46.5%).
- Chirality Verification: Explicit geometric reasoning allowed MolScribe to predict chiral molecules significantly better than image-to-SMILES baselines. When chirality is ignored, the performance gap narrows (e.g., on Indigo, baseline accuracy rises from 94.1% to 96.3%), isolating MolScribe’s primary advantage to geometric reasoning for stereochemistry.
- Hand-Drawn Generalization: The model achieved 11.2% exact match accuracy on the DECIMER-HDM dataset, despite lacking hand-drawn images in the training set, with many errors bounded to a few atomic mismatches.
- Robustness: The model maintained high performance on perturbed images (rotation/shear), whereas rule-based systems degraded severely.
- Usability: The atom-level alignment allows for confidence visualization, and human evaluation showed it reduced digitization time from 137s to 20s per molecule.
Reproducibility Details
Data
The model was trained on a mix of synthetic and patent data with extensive dynamic augmentation:
| Purpose | Dataset | Size | Notes |
|---|---|---|---|
| Training | PubChem (Synthetic) | 1M | Molecules randomly sampled from PubChem and rendered via Indigo toolkit; includes atom coords. |
| Training | USPTO (Patents) | 680K | Patent data lacks exact atom coordinates; relative coordinates normalized from MOLfiles to image dimensions (often introduces coordinate shifts). |
Molecule Augmentation:
- Functional Groups: Randomly substituted using 53 common substitution rules (e.g., replacing substructures with “Et” or “Ph”).
- R-Groups: Randomly added using vocabulary:
[R, R1...R12, Ra, Rb, Rc, Rd, X, Y, Z, A, Ar]. - Styles: Random variation of aromaticity (circle vs. bonds) and explicit hydrogens.
Image Augmentation:
- Rendering: Randomized font (Arial, Times, Courier, Helvetica), line width, and label modes during synthetic generation.
- Perturbations: Applied rotation ($\pm 90^{\circ}$), cropping ($1\%$), padding ($40\%$), downscaling, blurring, and Salt-and-Pepper/Gaussian noise.
Preprocessing: Input images are resized to $384 \x384$.
Algorithms
- Atom Prediction (Pix2Seq-style):
- The model generates a sequence of tokens: $S^A = [l_1, \hat{x}_1, \hat{y}_1, \dots, l_n, \hat{x}_n, \hat{y}_n]$.
- Discretization: Coordinates are binned into integer tokens ($n_{bins} = 64$).
- Tokenizer: Atom-wise tokenizer splits SMILES into atoms; non-atom tokens (parentheses, digits) are kept to help structure learning.
- Bond Prediction:
- Format: Pairwise classification for every pair of predicted atoms.
- Symmetry: For symmetric bonds (single/double), the probability is averaged as: $$ \hat{P}(b_{i,j} = t) = \frac{1}{2} \big( P(b_{i,j} = t) + P(b_{j,i} = t) \big) $$ For wedges, directional logic strictly applies instead.
- Abbreviation Expansion (Algorithm 1):
- A greedy algorithm connects atoms within an expanded abbreviation (e.g., “COOH”) until valences are full, avoiding the need for a fixed dictionary.
- Carbon Chains: Splits condensed chains like $C_aX_b$ into explicit sequences ($CX_q…CX_{q+r}$).
- Nested Formulas: Recursively parses nested structures like $N(CH_3)_2$ by treating them as superatoms attached to the current backbone.
- Valence Handling: Iterates through common valences first to resolve ambiguities.
Models
The architecture is an encoder-decoder with a classification head:
- Encoder: Swin Transformer (Swin-B), pre-trained on ImageNet-22K (88M params).
- Decoder: 6-layer Transformer, 8 heads, hidden dimension 256.
- Bond Predictor: 2-layer MLP (Feedforward) with ReLU, taking concatenated atom hidden states as input.
- Training: Teacher forcing, Cross-Entropy Loss, Batch size 128, 30 epochs.
Evaluation
Metric: Exact Match of Canonical SMILES.
- Stereochemistry: Must match tetrahedral chirality; cis-trans ignored.
- R-groups: Replaced with wildcards
*or[d*]for evaluation.
Hardware
- Compute: Training performed on Linux server with 96 CPUs and 500GB RAM.
- GPUs: 4x NVIDIA A100 GPUs.
- Training Time: Unspecified; comparative models on large datasets took “more than one day”.
- Inference: Requires autoregressive decoding for atoms, followed by a single forward pass for bonds.
Citation
@article{qianMolScribeRobustMolecular2023,
title = {{{MolScribe}}: {{Robust Molecular Structure Recognition}} with {{Image-To-Graph Generation}}},
shorttitle = {{{MolScribe}}},
author = {Qian, Yujie and Guo, Jiang and Tu, Zhengkai and Li, Zhening and Coley, Connor W. and Barzilay, Regina},
year = 2023,
month = apr,
journal = {Journal of Chemical Information and Modeling},
volume = {63},
number = {7},
pages = {1925--1934},
doi = {10.1021/acs.jcim.2c01480},
url = {https://pubs.acs.org/doi/10.1021/acs.jcim.2c01480}
}