Paper Information
Title: MolGrapher: Graph-based Visual Recognition of Chemical Structures
Authors: Lucas Morin, Martin Danelljan, Maria Isabel Agea, Ahmed Nassar, Valery Weber, Ingmar Meijer, Peter Staar, Fisher Yu
Citation: Morin, L., Danelljan, M., Agea, M. I., Nassar, A., Weber, V., Meijer, I., Staar, P., & Yu, F. (2023). MolGrapher: Graph-based Visual Recognition of Chemical Structures. Proceedings of the IEEE/CVF International Conference on Computer Vision (ICCV), 19552-19561.
Publication: ICCV 2023
Links:
1. Contribution / Type
This is primarily a Methodological paper that proposes a novel neural architecture (MolGrapher), shifting the paradigm of Optical Chemical Structure Recognition (OCSR) from image captioning back to graph reconstruction. It also has a significant Resource component, releasing a synthetic data generation pipeline and a new large-scale benchmark (USPTO-30K) to address the scarcity of annotated real-world data.
2. Motivation
The automatic analysis of chemical literature is critical for accelerating drug and material discovery, but much of this information is locked in 2D images of molecular structures.
- Problem: Existing rule-based methods are rigid, while recent deep learning methods based on “image captioning” (predicting SMILES strings) struggle with complex molecules and fail to exploit the natural graph structure of molecules.
- Gap: There is a lack of diverse, annotated real-world training data, and captioning models suffer from “hallucinations” where they predict valid SMILES that do not match the image.
3. Novelty / Core Innovation
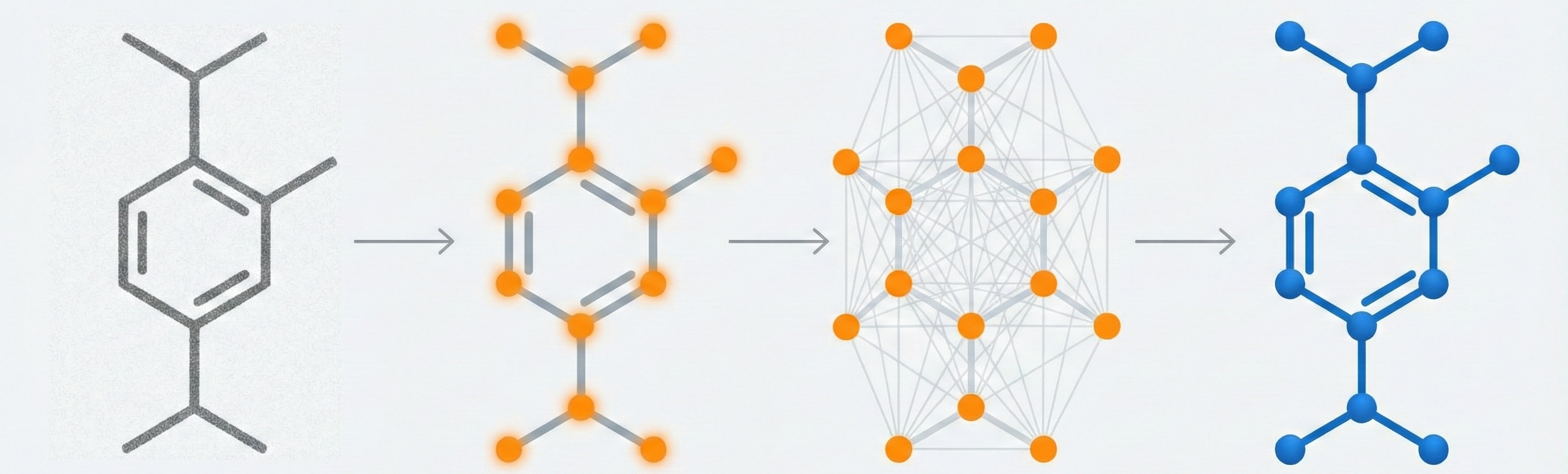
MolGrapher introduces a graph-based deep learning pipeline that explicitly models the molecule’s geometry and topology.
- Supergraph Concept: It first detects all atom keypoints and builds a “supergraph” of all plausible bonds.
- Hybrid Approach: It combines a ResNet-based keypoint detector with a Graph Neural Network (GNN) that classifies nodes (atoms) and edges (bonds) within the supergraph context.
- Synthetic Pipeline: A robust data generation pipeline that renders molecules with varying styles (fonts, bond widths) and augmentations to simulate real document noise.
At the core of the Keypoint Detector’s performance is the Weight-Adaptive Heatmap Regression (WAHR) loss. Since pixels without an atom drastically outnumber pixels containing an atom, WAHR loss is designed to counter the class imbalance. For ground-truth heatmap $y$ and prediction $p$:
$$ L_{WAHR}(p, y) = \sum_i \alpha_y (p_i - y_i)^2 $$
where $\alpha_y$ dynamically down-weights easily classified background pixels.
4. Methodology & Experiments
The authors evaluated MolGrapher against both rule-based (OSRA, MolVec) and deep learning baselines (DECIMER, Img2Mol, Image2Graph).
- Benchmarks: Evaluated on standard datasets: USPTO, Maybridge UoB, CLEF-2012, and JPO.
- New Benchmark: Introduced and tested on USPTO-30K, split into clean, abbreviated, and large molecule subsets.
- Ablations: Analyzed the impact of synthetic augmentations, keypoint loss functions, supergraph connectivity radius, and GNN layers.
- Robustness: Tested on perturbed images (rotations, shearing) to mimic scanned patent quality.
The key mathematical formulation in the model involves the node context updates in the GNN mechanism. Messages $m_{j\to i}$ from neighboring nodes are aggregated to form a topological update $\Delta x_i$, which is then combined with visual updates.
5. Results & Conclusions
MolGrapher achieved state-of-the-art performance, significantly outperforming image captioning methods on standard benchmarks.
- Accuracy: It achieved 91.5% accuracy on USPTO compared to 61.0% for DECIMER 2.0 (the next best synthetic-only method).
- Large Molecules: It demonstrated superior scaling, correctly recognizing large molecules (USPTO-10K-L) where image captioning methods like Img2Mol failed completely (0.0% accuracy).
- Generalization: The method proved robust to image perturbations and style variations without requiring fine-tuning on real data. Important caveats: The paper acknowledges MolGrapher’s sensitivity to disconnected structures and lack of support for stereochemistry mapping.
Reproducibility Details
Data
The model relies on synthetic data for training due to the scarcity of annotated real-world images.
| Purpose | Dataset | Size | Notes |
|---|---|---|---|
| Training | Synthetic Data | 300,000 images | Generated from PubChem SMILES using RDKit. Augmentations include pepper patches, random lines, and variable bond styles. |
| Evaluation | USPTO-30K | 30,000 images | Created by authors from USPTO patents (2001-2020). Subsets: 10K clean, 10K abbreviated, 10K large (>70 atoms). |
| Evaluation | Standard Benchmarks | Various | USPTO (5,719), Maybridge UoB (5,740), CLEF-2012 (992), JPO (450). |
Algorithms
The pipeline consists of three distinct algorithmic stages:
Keypoint Detection:
- Predicts a heatmap of atom locations using a CNN.
- Thresholds heatmaps at the bottom 10th percentile and uses a $5\times5$ window for local maxima.
- Uses Weight-Adaptive Heatmap Regression (WAHR) loss to handle class imbalance (background vs. atoms).
Supergraph Construction:
- Connects every detected keypoint to neighbors within a radius of $3 \times$ the estimated bond length.
- Prunes edges with no filled pixels or if obstructed by a third keypoint.
- Keeps a maximum of 6 bond candidates per atom.
Superatom Recognition:
- Detects “superatom” nodes (abbreviations like
COOH). - Uses PP-OCR to transcribe the text at these node locations.
- Detects “superatom” nodes (abbreviations like
Models
The architecture utilizes standard backbones tailored for specific sub-tasks:
- Keypoint Detector: ResNet-18 backbone with $8\times$ dilation to preserve spatial resolution.
- Node Classifier: ResNet-50 backbone with $2\times$ dilation for extracting visual features at node locations.
- Graph Neural Network: A custom GNN that updates node embeddings based on visual features and neighborhood context. The initial node embedding combines the visual feature vector $v_i$ and a learnable type encoding $w_{t_i}$.
- Readout: MLPs classify nodes into atom types (e.g., C, O, N) and bond types (Single, Double, None).
Evaluation
Accuracy is defined strictly: the predicted molecule must have an identical InChI string to the ground truth. Stereochemistry and Markush structures are excluded from evaluation.
| Metric | Dataset | MolGrapher Score | SOTA Baseline (Synthetic) | Notes |
|---|---|---|---|---|
| Accuracy | USPTO | 91.5% | 61.0% (DECIMER 2.0) | Full USPTO benchmark |
| Accuracy | USPTO-10K-L | 31.4% | 0.0% (Img2Mol) | Large molecules (>70 atoms) |
| Accuracy | JPO | 67.5% | 64.0% (DECIMER 2.0) | Challenging, low-quality images |
Hardware
- GPUs: Trained on 3 NVIDIA A100 GPUs.
- Training Time: 20 epochs.
- Optimization: ADAM optimizer, learning rate 0.0001, decayed by 0.8 after 5000 iterations.
- Loss Weighting: Atom classifier loss weighted by 1; bond classifier loss weighted by 3.
Citation
@inproceedings{morinMolGrapherGraphbasedVisual2023,
title = {{{MolGrapher}}: {{Graph-based Visual Recognition}} of {{Chemical Structures}}},
shorttitle = {{{MolGrapher}}},
booktitle = {Proceedings of the {{IEEE}}/{{CVF International Conference}} on {{Computer Vision}}},
author = {Morin, Lucas and Danelljan, Martin and Agea, Maria Isabel and Nassar, Ahmed and Weber, Valery and Meijer, Ingmar and Staar, Peter and Yu, Fisher},
year = {2023},
pages = {19552--19561},
doi = {10.1109/ICCV51070.2023.01793},
urldate = {2025-10-18},
langid = {english}
}