Paper Information
Citation: Hu, F., He, E., & Verspoor, K. (2026). AdaptMol: Domain Adaptation for Molecular Image Recognition with Limited Supervision. Research Square preprint. https://doi.org/10.21203/rs.3.rs-8365561/v1
Publication: Research Square preprint, February 2026
Additional Resources:
Bridging the Synthetic-to-Real Gap in Graph-Based OCSR
Most OCSR methods are trained on synthetic molecular images and evaluated on high-quality literature figures, both exhibiting relatively uniform styles. Hand-drawn molecules represent a particularly challenging domain with irregular bond lengths, variable stroke widths, and inconsistent atom symbols. Prior graph reconstruction methods like MolScribe and MolGrapher drop below 15% accuracy on hand-drawn images, despite achieving over 65% on literature datasets.
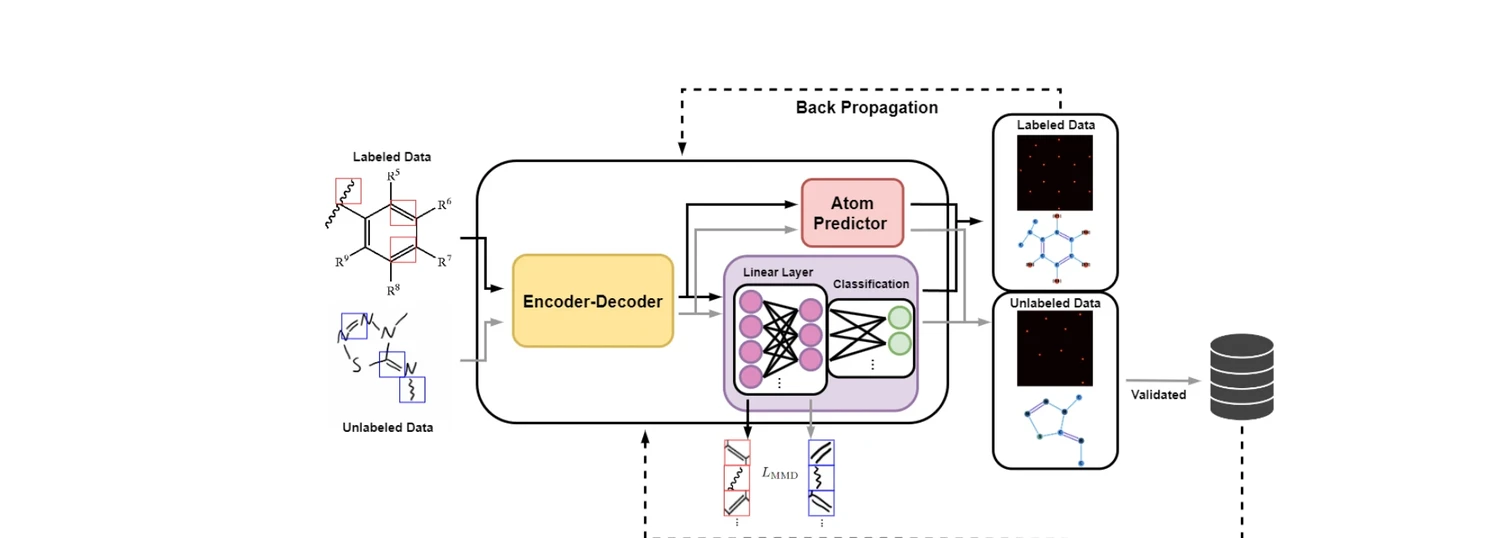
AdaptMol addresses this with a three-stage pipeline that enables effective transfer from synthetic to real-world data without requiring graph annotations in the target domain:
- Base model training on synthetic data with comprehensive augmentation and dual position representation
- MMD alignment of bond-level features between source and target domains
- Self-training with SMILES-validated pseudo-labels on unlabeled target images
End-to-End Graph Reconstruction Architecture
AdaptMol builds on MolScribe’s architecture, using a Swin Transformer base encoder ($384 \times 384$ input) with a 6-layer Transformer decoder (8 heads, hidden dim 256). The model jointly predicts atoms and bonds:
Atom prediction follows the Pix2Seq approach, autoregressively generating a sequence of atom tokens:
$$S_N = [l_1, x_1, y_1, l_2, x_2, y_2, \dots, l_n, x_n, y_n]$$
where $l_i$ is the atom label and $(x_i, y_i)$ are discretized coordinate bin indices.
Dual position representation adds a 2D spatial heatmap on top of token-based coordinate prediction. The heatmap aggregates joint spatial distributions of all atoms:
$$\mathbf{H} = \text{Upsample}\left(\sum_{i=1}^{n} P_y^{(i)} \otimes P_x^{(i)}\right)$$
where $P_x^{(i)}$ and $P_y^{(i)}$ are coordinate probability distributions from the softmax logits. During training, this heatmap is supervised with Gaussian kernels at ground-truth atom positions. This reduces false positive atom predictions substantially (from 356 to 33 false positives at IoU 0.05).
Bond prediction extracts atom-level features from decoder hidden states and enriches them with encoder visual features via multi-head attention with a learnable residual weight $\alpha$:
$$\mathbf{F}{\text{enriched}} = \text{LayerNorm}(\mathbf{F}{\text{atom}} + \alpha \cdot \text{MHA}(\mathbf{F}{\text{atom}}, \mathbf{E}{\text{vis}}))$$
A feed-forward network then predicts bond types between all atom pairs.
Bond-Level Domain Adaptation via MMD
The key insight is that bond features are domain-invariant: they encode structural relationships (single, double, triple, aromatic) independent of visual style. Atom-level alignment is problematic due to class imbalance (carbon dominates), multi-token spanning (functional groups), and position-dependent features.
AdaptMol aligns bond-level feature distributions via class-conditional Maximum Mean Discrepancy:
$$L_{\text{MMD}} = \frac{1}{|\mathcal{C}’|} \sum_{c \in \mathcal{C}’} MMD(F_c^{\text{src}}, F_c^{\text{tgt}})$$
where $\mathcal{C}’$ contains classes with sufficient samples in both domains. Confidence-based filtering retains only high-confidence predictions (confidence > 0.95, entropy < 0.1) for alignment, tightening to 0.98 and 0.05 after the first epoch. Progressive loss weighting follows a schedule of 0.1 (epoch 0), 0.075 (epoch 1), and 0.05 thereafter.
An important side effect: MMD alignment improves inter-class bond discrimination, reducing confusion between visually similar bond types (e.g., jagged double bonds vs. aromatic bonds).
Self-Training with SMILES Validation
After MMD alignment, the model generates predictions on unlabeled target images. Predicted molecular graphs are converted to SMILES and validated against ground-truth SMILES annotations. Only exact matches are retained as pseudo-labels, providing complete graph supervision (atom coordinates, element types, bond types) that was previously unavailable in the target domain.
This approach is far more data-efficient than alternatives: AdaptMol uses only 4,080 real hand-drawn images vs. DECIMER-Handdraw’s 38 million synthetic hand-drawn images.
Comprehensive Data Augmentation
Two categories of augmentation are applied during synthetic data generation:
- Structure-rendering augmentation: Functional group abbreviation substitution, bond type conversions (single to wavy/aromatic, Kekule to aromatic rings), R-group insertion, and rendering parameter randomization (font family/size, bond width/spacing)
- Image-level augmentation: Geometric operations, quality degradation, layout variations, and chemical document artifacts (caption injection, arrows, marginal annotations)
Structure-rendering augmentation provides the larger benefit, contributing ~20% accuracy improvement on JPO and ~30% on ACS benchmarks.
Results
Hand-Drawn Molecule Recognition
| Method | DECIMER test (Acc) | ChemPix (Acc) |
|---|---|---|
| AdaptMol | 82.6 | 60.5 |
| DECIMER v2.2 | 71.9 | 51.4 |
| AtomLenz | 30.0 | 48.4 |
| MolScribe | 10.1 | 26.1 |
| MolGrapher | 10.7 | 14.5 |
Literature and Synthetic Benchmarks
AdaptMol achieves state-of-the-art on 4 of 6 literature benchmarks:
| Dataset | AdaptMol | MolScribe | MolGrapher | DECIMER v2.2 |
|---|---|---|---|---|
| CLEF | 92.7 | 87.5 | 57.2 | 77.7 |
| JPO | 88.2 | 78.8 | 73.0 | 75.7 |
| UOB | 89.3 | 88.2 | 85.1 | 87.2 |
| ACS | 75.5 | 72.8 | 41.0 | 37.7 |
| USPTO | 90.9 | 92.6 | 74.9 | 59.6 |
| Staker | 84.0 | 84.4 | 0.0 | 66.3 |
MolScribe edges out on USPTO and Staker. The authors attribute this to MolScribe directly training on all 680K USPTO samples, which may cause it to specialize to that distribution.
Pipeline Ablation
| Configuration | Hand-drawn | ChemDraw | JPO |
|---|---|---|---|
| Base model | 10.4 | 92.3 | 82.7 |
| + Font augmentation | 30.2 | 92.5 | 82.8 |
| + Font aug + MMD | 42.1 | 94.0 | 83.0 |
| + Font aug + MMD + Self-training | 82.6 | 95.9 | 88.2 |
Each component contributes meaningfully: font augmentation (+19.8), MMD alignment (+11.9), and self-training (+40.5) on hand-drawn accuracy.
Reproducibility
| Artifact | Type | License | Notes |
|---|---|---|---|
| AdaptMol Code | Code | MIT | Official implementation |
| Model + Data | Model/Dataset | MIT | Pretrained checkpoint and datasets |
Training uses 2 NVIDIA A100 GPUs (40GB each). Base model trains for 30 epochs on 1M synthetic samples. Domain adaptation involves 3 steps: USPTO self-training (3 iterations of 3 epochs), MMD alignment on hand-drawn data (5 epochs), and hand-drawn self-training (5 iterations).
Limitations
- Sequence length constraints prevent accurate prediction of very large molecules (>120 atoms), where resizing causes significant information loss
- Cannot recognize Markush structures with repeating unit notation (parentheses/brackets), as synthetic training data lacks such cases
- Stereochemistry information is lost when stereo bonds connect to abbreviated functional groups due to RDKit post-processing limitations
- The retrained baseline (30 epochs from scratch on synthetic + pseudo-labels) achieves higher hand-drawn accuracy (87.2%) but at the cost of cross-domain robustness on literature benchmarks