Deep learning OCSR methods that predict molecular graph structure directly from images, detecting atoms and bonds as nodes and edges.
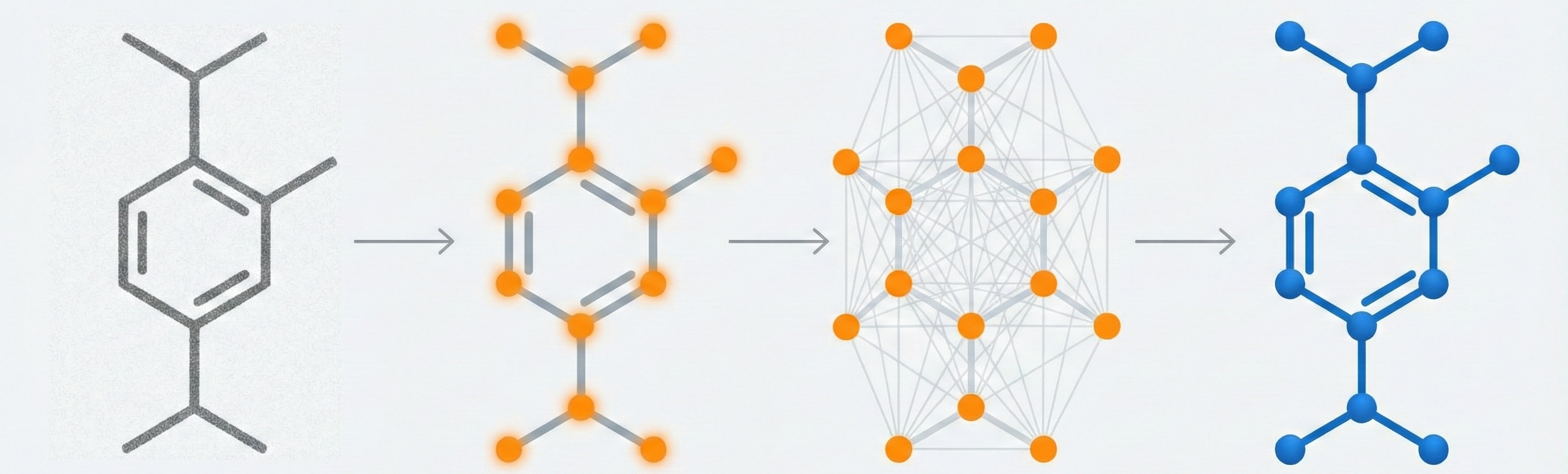
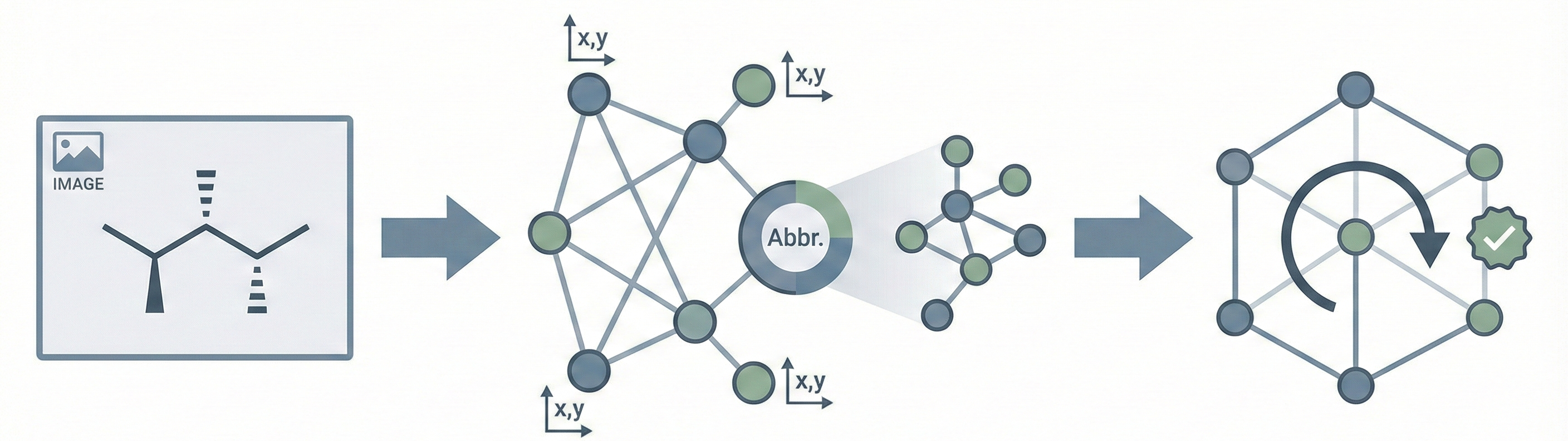
Image-to-graph methods bypass string representations entirely, predicting the molecular graph (atoms as nodes, bonds as edges) directly from the input image. This family includes segmentation-based approaches like ChemGrapher and Staker et al.’s U-Net pipeline, keypoint-detection architectures like ABC-Net, and joint atom-bond-coordinate predictors like MolScribe. By reasoning about spatial structure rather than linearizing it, these models tend to handle stereochemistry and abbreviated groups more naturally than sequence-based alternatives. Full-pipeline systems like MolMiner and MolMole extend the approach to page-level chemical extraction from documents.
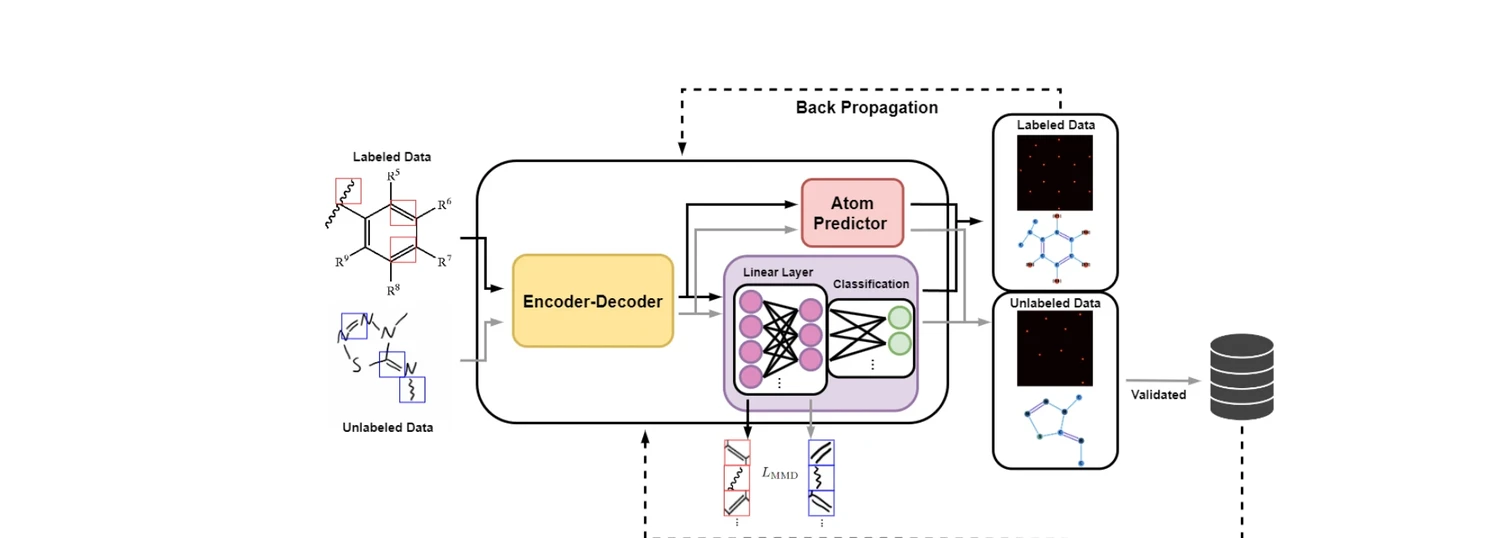
AdaptMol: Domain Adaptation for Molecular OCSR (2026)
AdaptMol combines an end-to-end graph reconstruction model with unsupervised domain adaptation via class-conditional MMD on bond features and SMILES-validated self-training. Achieves 82.6% accuracy on hand-drawn molecules (10.7 points above prior best) while maintaining state-of-the-art results on four literature benchmarks, using only 4,080 real hand-drawn images for adaptation.
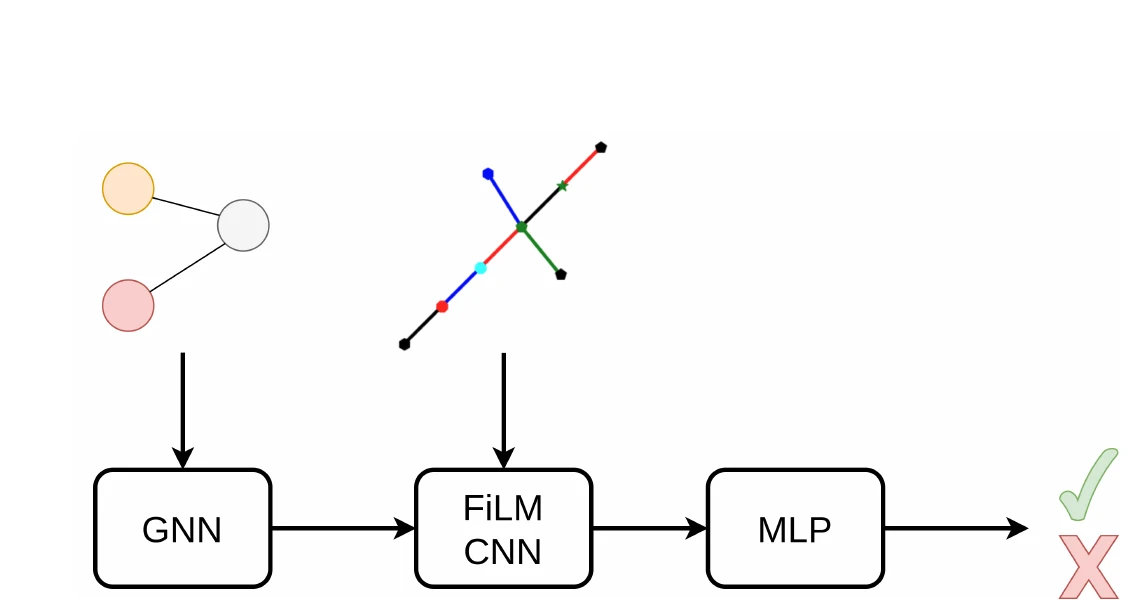
GraSP: Graph Recognition via Subgraph Prediction (2026)
GraSP introduces a general framework for recognizing graphs in images by framing it as sequential subgraph prediction with a binary classifier. A GNN conditions a CNN via FiLM layers to predict whether a candidate graph is a subgraph of the target. Applied to OCSR on QM9, GraSP achieves 67.5% accuracy with no domain-specific modifications.
MolGrapher: Graph-based Chemical Structure Recognition
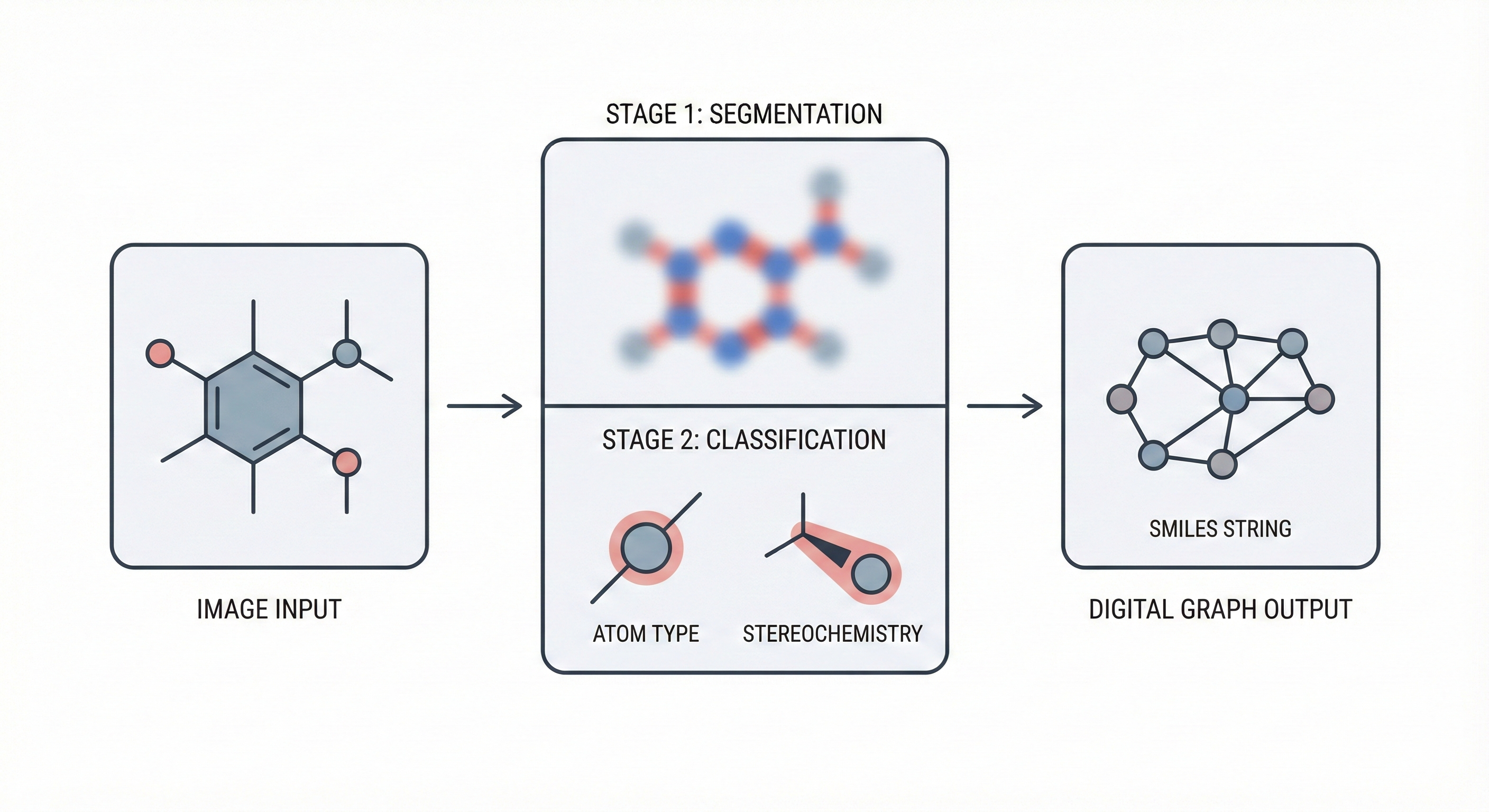
MolGrapher introduces a three-stage pipeline (keypoint detection, supergraph construction, GNN classification) for recognizing chemical structures from images. It achieves 91.5% accuracy on USPTO by treating molecules as graphs, and introduces the USPTO-30K benchmark.
MolMole: Unified Vision Pipeline for Molecule Mining
MolMole unifies molecule detection, reaction parsing, and structure recognition into a single vision-based pipeline, achieving top performance on a newly introduced 550-page benchmark by processing full documents without external layout parsers.
MolScribe reformulates molecular recognition as an image-to-graph generation task, explicitly predicting atom coordinates and bonds to better handle stereochemistry and abbreviated structures compared to image-to-SMILES baselines.
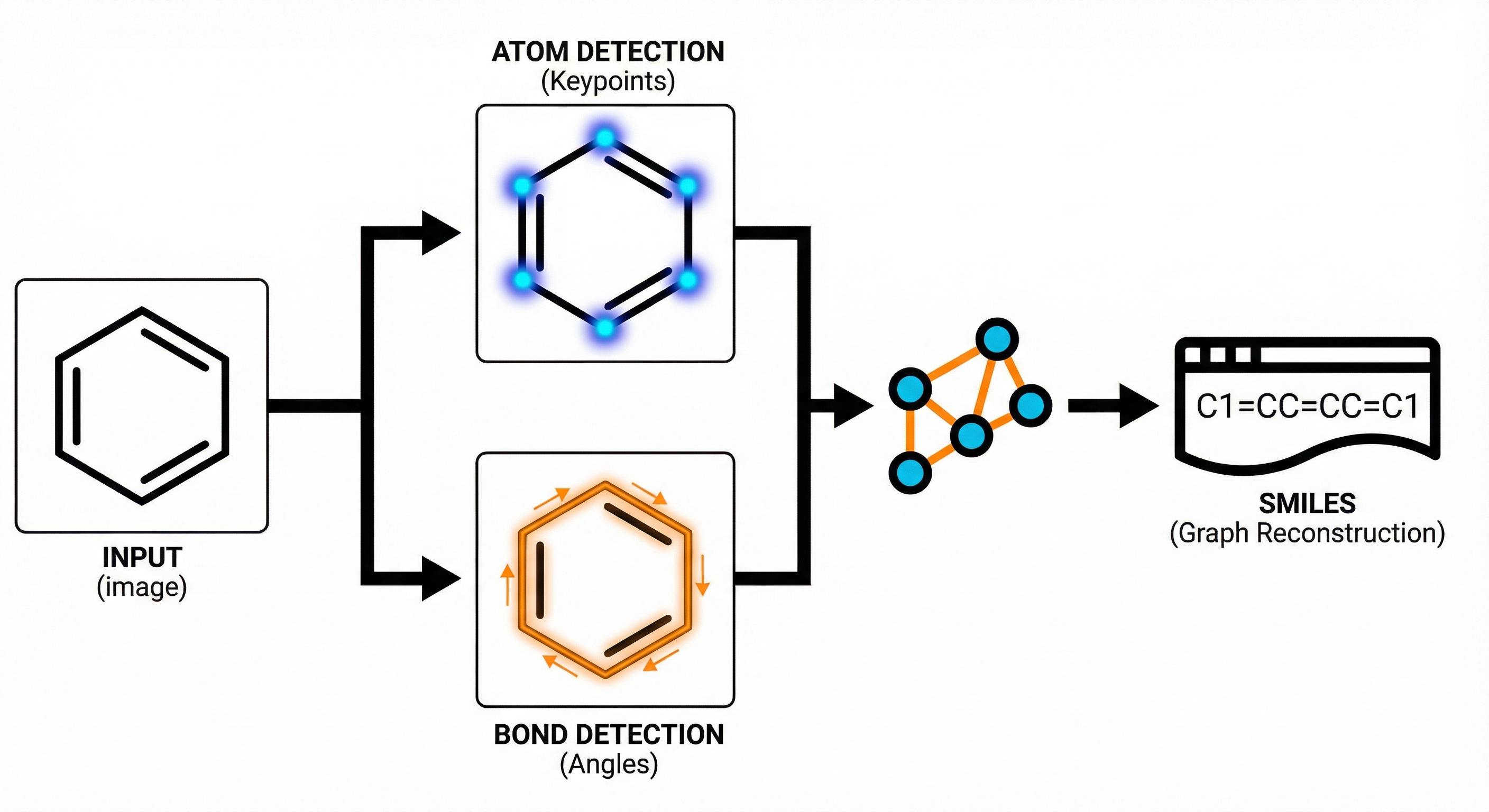
ABC-Net reformulates molecular image recognition as a keypoint detection problem. By predicting atom/bond centers and properties via a single Fully Convolutional Network, it achieves >94% accuracy with high data efficiency.
Image-to-Graph Transformers for Chemical Structures
This paper proposes an end-to-end deep learning architecture that translates chemical images directly into molecular graphs using a ResNet-Transformer encoder and a graph-aware decoder. It addresses the limitations of SMILES-based approaches by effectively handling non-atomic symbols (abbreviations) and varying drawing styles found in scientific literature.
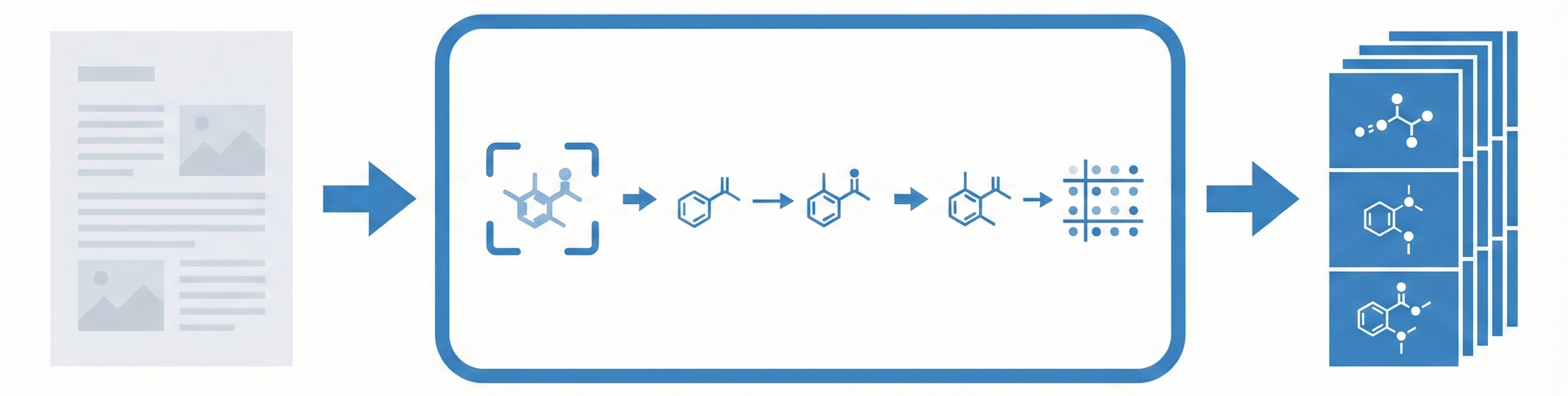
MolMiner: Deep Learning OCSR with YOLOv5 Detection
MolMiner replaces traditional rule-based vectorization with a deep learning object detection pipeline (YOLOv5) to extract chemical structures from PDFs. It outperforms open-source baselines on four benchmarks and introduces a new real-world dataset of 3,040 images.
ChemGrapher: Deep Learning for Chemical Graph OCSR
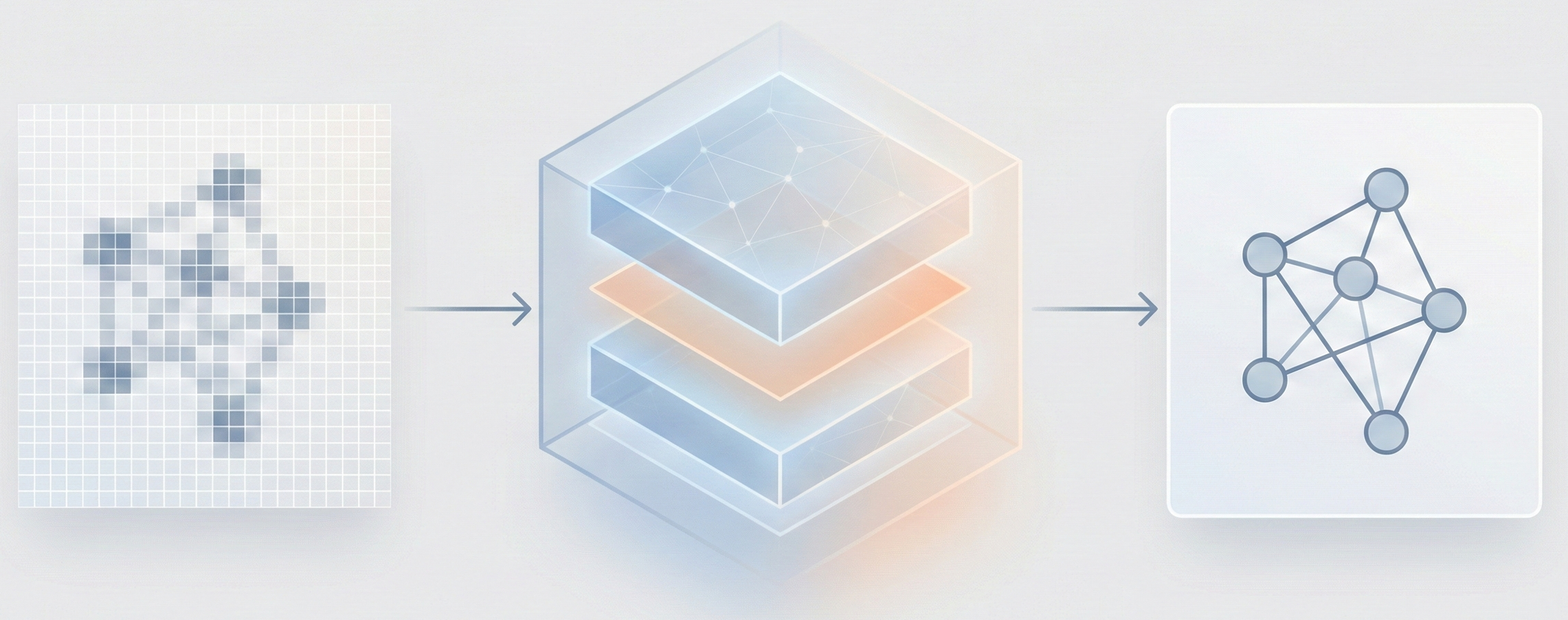
ChemGrapher replaces rule-based chemical OCR with a deep learning pipeline using semantic segmentation to identify atom and bond candidates, followed by specialized classification networks to resolve stereochemistry and bond multiplicity, reducing error rates compared to OSRA across all tested styles.