Dual Contribution: Method and Data Resource
The paper proposes a novel architecture (AtomLenz) and training framework (ProbKT* + Edit-Correction) to solve the problem of Optical Chemical Structure Recognition (OCSR) in data-sparse domains. As a secondary projection, it serves as a Resource paper by releasing a curated, relabeled dataset of hand-drawn molecules explicitly bound to atom-level bounding box annotations.
Overcoming Annotation Bottlenecks in OCSR
Optical Chemical Structure Recognition (OCSR) is critical for digitizing chemical literature and lab notes. However, existing methods face three main limitations:
- Generalization Limits: They struggle with sparse or stylistically unique domains, such as hand-drawn images, where massive datasets for pretraining are unavailable.
- Annotation Cost: “Atom-level” methods (which detect individual atoms and bonds) require expensive bounding box annotations, which are rarely available for real-world sketch data.
- Lack of Interpretability/Localization: Pure “Image-to-SMILES” models (like DECIMER) work well but fail to localize the atoms or bonds in the original image, limiting human-in-the-loop review and mechanistic interpretability.
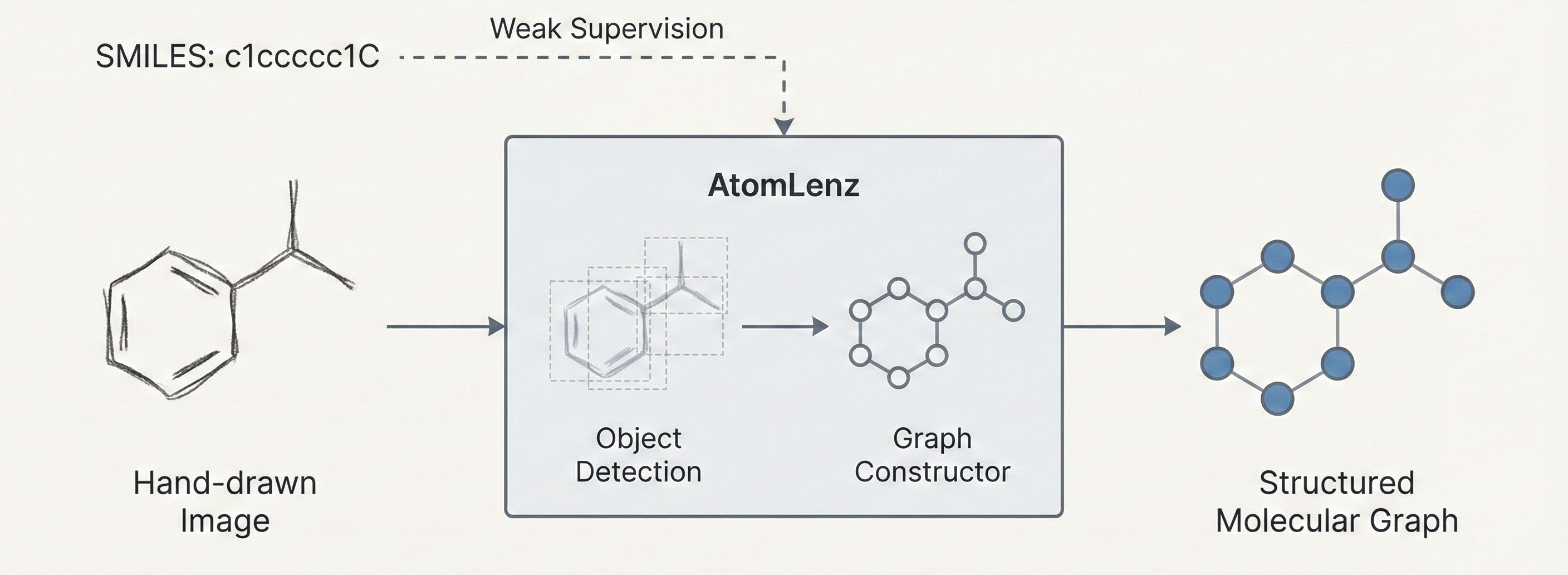
AtomLenz, ProbKT*, and Graph Edit-Correction
The core contribution is AtomLenz, an OCSR framework that achieves atom-level entity detection using only SMILES supervision on target domains. The authors construct an explicit object detection pipeline using Faster R-CNN trained via a composite multi-task loss. The objective aims to optimize a multi-class log loss $L_{cls}$ for predicted class $\hat{c}$ and a regression loss $L_{reg}$ for predicted bounding box coordinates $\hat{b}$:
$$ \mathcal{L} = L_{cls}(c, \hat{c}) + L_{reg}(b, \hat{b}) $$
To bridge the gap between image inputs and the weakly supervised SMILES labels, the system leverages:
- ProbKT (Probabilistic Knowledge Transfer):* Uses probabilistic logic and Hungarian matching to align predicted objects with the “ground truth” derived from the SMILES strings, enabling backpropagation without explicitly bounding boxes.
- Graph Edit-Correction: Generates pseudo-labels by solving for the minimum graph edit distance between the model’s predicted output graph and the ground-truth SMILES graph, which forces fine-tuning on less frequent atom types.
- ChemExpert: A chemically sound ensemble strategy that cascades predictions from multiple models (e.g., passing through DECIMER, then AtomLenz), halting at the first output that clears basic RDKit chemical validity checks.
Data Efficiency and Domain Adaptation Experiments
The authors evaluated the model specifically on domain adaptation and sample efficiency, treating hand-drawn molecules as the primary low-data target distribution:
- Pretraining: Initially trained on ~214k synthetic images from ChEMBL explicitly labeled with bounding boxes (generated via RDKit).
- Target Domain Adaptation: Fine-tuned on the Brinkhaus hand-drawn dataset (4,070 images) using purely SMILES supervision.
- Evaluation Sets:
- Hand-drawn test set: 1,018 images.
- ChemPix: 614 out-of-domain hand-drawn images.
- Atom Localization set: 1,000 synthetic images to evaluate precise bounding box capabilities.
- Baselines: Compared against leading OCSR methods, including DECIMER (v2.2), Img2Mol, MolScribe, ChemGrapher, and OSRA.
State-of-the-Art Ensembles vs. Standalone Limitations
- SOTA Ensemble Performance: The ChemExpert module (combining AtomLenz and DECIMER) achieved state-of-the-art accuracy on both hand-drawn (63.5%) and ChemPix (51.8%) test sets.
- Data Efficiency under Bottleneck Regimes: AtomLenz effectively bypassed the massive data constraints of competing models. When strictly limited to a training set of 4,000 samples from scratch, AtomLenz achieved 33.8% exact accuracy, outperforming baselines like Img2Mol (0.0%), MolScribe (1.3%), and DECIMER (0.1%), illustrating its tremendous sample efficiency.
- Localization Success: The base framework confidently maintained strong localization capabilities (mAP 0.801) absent from end-to-end transformers like DECIMER.
- Methodological Tradeoffs: While AtomLenz is highly sample efficient, its standalone performance when fine-tuned on the target domain (33.8% accuracy) underperforms fine-tuned models trained on larger datasets like DECIMER (62.2% accuracy). AtomLenz achieves state-of-the-art results primarily when deployed as part of the ChemExpert ensemble alongside DECIMER, functioning effectively as a synergistic error-correction mechanism.
Reproducibility Details
Artifacts
| Artifact | Type | License | Notes |
|---|---|---|---|
| Official Repository (AtomLenz) | Code | MIT | Complete pipeline for AtomLenz, ProbKT*, and Graph Edit-Correction. |
| Pre-trained Models | Model | MIT | Downloadable weights for Faster R-CNN detection backbones. |
| Hand-drawn Dataset (Brinkhaus) | Dataset | Unknown | Images and annotations used for target domain fine-tuning and evaluation. |
| AtomLenz Web Demo | Other | Unknown | Interactive Hugging Face space for testing model inference. |
Data
The study utilizes a mix of large synthetic datasets and smaller curated hand-drawn datasets.
| Purpose | Dataset | Size | Notes |
|---|---|---|---|
| Pretraining | Synthetic ChEMBL | ~214,000 | Generated via RDKit/Indigo. Annotated with atoms, bonds, charges, stereocenters. |
| Fine-tuning | Hand-drawn (Brinkhaus) | 4,070 | Used for weakly supervised adaptation (SMILES only). |
| Evaluation | Hand-drawn Test | 1,018 | |
| Evaluation | ChemPix | 614 | Out-of-distribution hand-drawn images. |
| Evaluation | Atom Localization | 1,000 | Synthetic images with ground truth bounding boxes. |
Algorithms
- Molecular Graph Constructor (Algorithm 1): A rule-based system to assemble the graph from detected objects:
- Filtering: Removes overlapping atom boxes (IoU threshold).
- Node Creation: Merges charge and stereocenter objects with the nearest atom objects.
- Edge Creation: Iterates over bond objects; if a bond overlaps with exactly two atoms, an edge is added. If >2, it selects the most probable pair.
- Validation: Checks valency constraints; removes bonds iteratively if constraints are violated.
- Weakly Supervised Training:
- ProbKT*: Uses Hungarian matching to align predicted objects with the “ground truth” implied by the SMILES string, allowing backpropagation without explicit boxes.
- Graph Edit-Correction: Computes the Minimum Graph Edit Distance between the predicted graph and the true SMILES graph to generate pseudo-labels for retraining.
Models
- Object Detection Backbone: Faster R-CNN.
- Four distinct models are trained for different entity types: Atoms ($O^a$), Bonds ($O^b$), Charges ($O^c$), and Stereocenters ($O^s$).
- Loss Function: Multi-task loss combining Multi-class Log Loss ($L_{cls}$) and Regression Loss ($L_{reg}$).
- ChemExpert: An ensemble wrapper that prioritizes models based on user preference (e.g., DECIMER first, then AtomLenz). It accepts the first prediction that passes RDKit chemical validity checks.
Evaluation
Primary metrics focused on structural correctness and localization accuracy.
| Metric | Value (Hand-drawn) | Baseline (DECIMER FT) | Notes |
|---|---|---|---|
| Accuracy (T=1) | 33.8% (AtomLenz+EditKT*) | 62.2% | Exact ECFP6 fingerprint match. |
| Tanimoto Sim. | 0.484 | 0.727 | Average similarity. |
| mAP | 0.801 | N/A | Localization accuracy (IoU 0.05-0.35). |
| Ensemble Acc. | 63.5% | 62.2% | ChemExpert (DECIMER + AtomLenz). |
Hardware
- Compute: Experiments utilized the Flemish Supercomputer Center (VSC) resources.
- Note: Specific GPU models (e.g., A100/V100) are not explicitly detailed in the text, but Faster R-CNN training is standard on consumer or enterprise GPUs.
Paper Information
Citation: Oldenhof, M., De Brouwer, E., Arany, A., & Moreau, Y. (2024). Atom-Level Optical Chemical Structure Recognition with Limited Supervision. arXiv preprint arXiv:2404.01743. https://doi.org/10.48550/arXiv.2404.01743
Publication venue/year: arXiv 2024
Additional Resources:
BibTeX:
@misc{oldenhofAtomLevelOpticalChemical2024,
title = {Atom-{{Level Optical Chemical Structure Recognition}} with {{Limited Supervision}}},
author = {Oldenhof, Martijn and Brouwer, Edward De and Arany, Adam and Moreau, Yves},
year = 2024,
month = apr,
number = {arXiv:2404.01743},
eprint = {2404.01743},
primaryclass = {cs},
publisher = {arXiv},
doi = {10.48550/arXiv.2404.01743},
urldate = {2025-10-25},
abstract = {Identifying the chemical structure from a graphical representation, or image, of a molecule is a challenging pattern recognition task that would greatly benefit drug development. Yet, existing methods for chemical structure recognition do not typically generalize well, and show diminished effectiveness when confronted with domains where data is sparse, or costly to generate, such as hand-drawn molecule images. To address this limitation, we propose a new chemical structure recognition tool that delivers state-of-the-art performance and can adapt to new domains with a limited number of data samples and supervision. Unlike previous approaches, our method provides atom-level localization, and can therefore segment the image into the different atoms and bonds. Our model is the first model to perform OCSR with atom-level entity detection with only SMILES supervision. Through rigorous and extensive benchmarking, we demonstrate the preeminence of our chemical structure recognition approach in terms of data efficiency, accuracy, and atom-level entity prediction.},
archiveprefix = {arXiv},
langid = {english},
keywords = {Computer Science - Computer Vision and Pattern Recognition},
file = {/Users/hunterheidenreich/Zotero/storage/4ILTDIFX/Oldenhof et al. - 2024 - Atom-Level Optical Chemical Structure Recognition with Limited Supervision.pdf}
}