A Fragment-Based Molecular Representation Method
This is a Method paper that proposes t-SMILES (tree-based SMILES), a framework for representing molecules as SMILES-type strings derived from fragment-based decompositions. The primary contribution is an encoding algorithm that converts fragmented molecular graphs into full binary trees (FBTs) and then traverses them breadth-first to produce linear strings. Three coding variants are introduced: TSSA (shared atom), TSDY (dummy atom without ID), and TSID (dummy atom with ID). The framework achieves 100% theoretical validity, higher novelty scores, and improved distribution-learning metrics compared to classical SMILES, DeepSMILES, and SELFIES across ChEMBL, ZINC, and QM9 benchmarks.
Why Fragment-Based Representations Matter for Molecular Generation
Classical SMILES encodes molecules via depth-first traversal of the molecular graph, requiring parentheses and ring identifiers to appear in matched pairs with deep nesting. When generative models (LSTM, Transformer) are trained on SMILES, they produce chemically invalid strings, particularly on small datasets, because they struggle to learn these long-range pairing constraints. DeepSMILES addresses some syntactical issues but still permits semantic violations (e.g., oxygen with three bonds). SELFIES guarantees 100% valid strings but at the cost of readability and, as the authors show, lower FCD scores indicating generated molecules diverge from the training distribution.
Fragment-based approaches reduce the search space compared to atom-level methods and can provide insights into molecular recognition (e.g., protein-ligand interactions). However, existing fragment-based deep learning methods rely on fixed dictionaries of candidate fragments, creating in-vocabulary/out-of-vocabulary problems and high-dimensional sparse representations. The encoding of fragments as SMILES-type strings, rather than dictionary IDs, had not been systematically explored before this work.
The authors draw on the observation that fragments in organic molecules follow a Zipf-like rank distribution similar to words in natural language, motivating the use of NLP techniques for fragment-based molecular modeling.
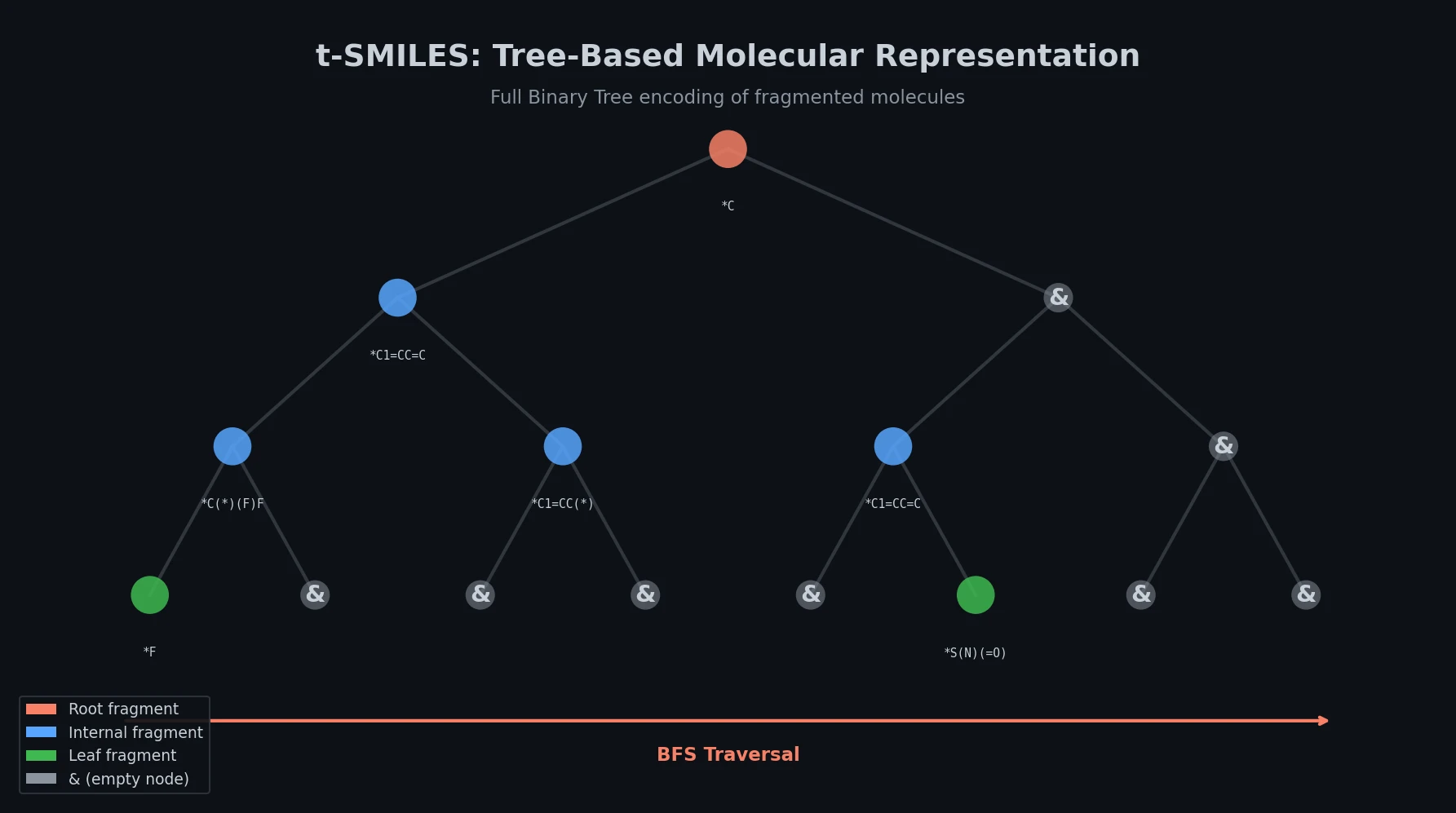
Core Innovation: Binary Tree Encoding of Fragmented Molecules
The t-SMILES algorithm proceeds in three steps:
- Fragmentation: A molecule is decomposed into valid chemical fragments using a chosen algorithm (JTVAE, BRICS, MMPA, or Scaffold), producing a fragmented molecular graph.
- Tree construction: The fragmented graph is converted into an Acyclic Molecular Tree (AMT), which is a reduced graph where nodes represent fragments and edges represent bonds between them. The AMT is then transformed into a Full Binary Tree (FBT), where every internal node has exactly two children.
- String generation: The FBT is traversed using breadth-first search (BFS) to produce the t-SMILES string.
The framework introduces only two new symbols beyond standard SMILES: & marks empty tree nodes (branch terminators providing global structural information), and ^ separates adjacent substructure segments (analogous to spaces between words in English).
Three Coding Variants
- TSSA (shared atom): Two fragments share a real atom at their connection point. Produces the highest novelty scores and is recommended for goal-directed tasks.
- TSDY (dummy atom, no ID): Uses dummy atoms (marked with
*) to indicate bonding points. Provides a balanced choice between novelty and distribution fidelity. - TSID (dummy atom with ID): Uses numbered dummy atoms (
[n*]) for unambiguous reconstruction. Produces the most faithful distribution reproduction and is recommended for distribution-learning tasks.
Structural Advantages
The key structural benefit is a dramatic reduction in nesting depth. For TSDY_M on ChEMBL, the proportion of tokens at nesting depth 0-1-2 increases from 68.0% (SMILES) to 99.3%, while depth 3-4-5 drops from 31.9% to 0.7%, and depth 6-11 drops from 0.1% to 0.0002%. The & symbol, which encodes molecular topology, does not need to appear in pairs (unlike parentheses in SMILES), and its high frequency means it does not create a scarcity problem for learning.
The framework also supports a multi-code system where classical SMILES can be integrated as a special case called TS_Vanilla, and multiple fragmentation-based codes can be combined into hybrid models.
Reconstruction and Data Augmentation
Molecules can be reconstructed from t-SMILES strings by reversing the process: rebuilding the FBT from the string, converting to AMT, and assembling fragments into a molecular graph. This reconstruction process can itself generate novel molecules without any model training by randomly assembling fragments. On ChEMBL, TSSA reconstruction achieves uniqueness above 0.98 and novelty above 0.68 for all four fragmentation algorithms, with 100% validity.
Data augmentation in t-SMILES operates at four levels: (1) different decomposition algorithms, (2) reconstruction, (3) enumeration of fragment strings, and (4) enumeration of FBTs. Unlike SMILES enumeration (which only produces different strings for the same molecule), t-SMILES reconstruction generates genuinely different molecules from the same fragment set.
Systematic Evaluation Across Multiple Benchmarks
All experiments use MolGPT (a Transformer-decoder model) as the primary generative model. Three types of metrics are employed: distribution-learning benchmarks, goal-directed benchmarks, and Wasserstein distance metrics for physicochemical properties.
Low-Resource Datasets (JNK3 and AID1706)
On JNK3 (923 active molecules), the authors investigate overfitting behavior across training epochs:
| Model | Valid | Novelty | FCD | Active Novel |
|---|---|---|---|---|
| SMILES [R200] | 0.795 | 0.120 | 0.584 | 0.072 |
| SMILES [R2000] | 1.000 | 0.001 | 0.765 | 0.004 |
| SELFIES [R200] | 1.000 | 0.238 | 0.544 | 0.148 |
| SELFIES [R2000] | 1.000 | 0.008 | 0.767 | 0.050 |
| TSSA_S [R300] | 1.000 | 0.833 | 0.564 | 0.582 |
| TSSA_S [R5000] | 1.000 | 0.817 | 0.608 | 0.564 |
| TF_TSSA_S [R5] | 1.000 | 0.932 | 0.483 | 0.710 |
| TSSA_S_Rec50 [R10] | 1.000 | 0.962 | 0.389 | 0.829 |
Key findings: SMILES and DeepSMILES novelty scores collapse to near zero after 200 epochs, while t-SMILES novelty stabilizes around 0.8. The highest active-novel score of 0.829 comes from t-SMILES with reconstruction-based data augmentation. Transfer learning with t-SMILES maintains novelty of 0.710 at 5 epochs versus 0.526 for SMILES, and at 100 epochs the gap widens dramatically (0.569 vs. 0.023).
Distribution Learning on ChEMBL
t-SMILES models outperform graph baselines (Graph MCTS, hG2G, MGM) and fragment-based methods (FASMIFRA). TSID_B and TSID_S achieve FCD scores of 0.909 while maintaining novelty of 0.941 and 0.933, surpassing SMILES (FCD 0.906, novelty 0.907) in both dimensions. TSDY and TSID models consistently outperform TSSA on distribution fidelity for larger molecules.
Goal-Directed Tasks on ChEMBL
On 20 GuacaMol subtasks, different fragmentation algorithms excel at different tasks. The goal-directed reconstruction algorithm significantly outperforms random reconstruction. On the Sitagliptin MPO task (T16.SMPO), the TSDY_M model with goal-directed reconstruction achieves a score of 0.930, compared to 0.598 for SMILES and 0.708 for CReM. On Valsartan SMARTS (T18.VS), t-SMILES models reach 0.997 versus 0.985 for SMILES.
Distribution Learning on ZINC and QM9
On ZINC, t-SMILES models significantly outperform existing fragment-based baselines (JTVAE, FragDgm). Seven t-SMILES models achieve both higher FCD and novelty scores than SELFIES. On QM9 (smaller molecules), all string-based models achieve high FCD scores (above 0.960), with t-SMILES performing better than existing string and graph approaches.
Physicochemical Properties
Across ChEMBL and ZINC, TSDY and TSID models capture physicochemical property distributions (MolWt, LogP, SAScore, N_Atoms, N_Rings, etc.) more faithfully than TSSA models. Multiple t-SMILES models outperform SMILES in more than four out of nine property categories. Baseline models hG2G and JTVAE show the weakest pattern learning, producing molecules with fewer atoms and rings than the training data.
Key Findings and Limitations
Main Results
- t-SMILES achieves 100% theoretical validity by fragmenting molecules into chemically valid pieces before encoding.
- The framework avoids the overfitting problem on low-resource datasets, maintaining stable novelty scores where SMILES, DeepSMILES, and SELFIES collapse.
- The multi-code system allows different coding algorithms to complement each other, with hybrid models accessing broader chemical space.
- Goal-directed reconstruction significantly outperforms all baselines on targeted optimization tasks.
- TSDY and TSID provide better distribution fidelity than TSSA on larger molecules, while TSSA excels at novelty generation for goal-directed tasks.
Limitations
The authors acknowledge several limitations:
- Whether the tree structure of t-SMILES can be effectively learned by Large Language Models remains unexplored.
- Only published fragmentation algorithms were tested; custom fragmentation schemes were not investigated.
- Experiments on more complex (larger) molecules were not performed.
- The reconstruction algorithm uses simple rules for fragment assembly; more sophisticated assembly methods (Monte Carlo tree search, CReM) could improve quality.
Future Directions
The authors suggest exploring advanced reconstruction and optimization algorithms, improved generative models, evolutionary techniques, and extending t-SMILES to property prediction, retrosynthesis, and reaction prediction tasks. The framework is also extensible to other string representations (t-DSMILES, t-SELFIES) by changing how fragments are encoded.
Reproducibility Details
Data
| Purpose | Dataset | Size | Notes |
|---|---|---|---|
| Low-resource evaluation | JNK3 | 923 active molecules | Kinase inhibitors |
| Low-resource evaluation | AID1706 | 329 active molecules | SARS 3CLPro inhibitors |
| Distribution learning | ChEMBL | Standard split | Large drug-like molecules |
| Distribution learning | ZINC | 250K subset | Medium drug-like molecules |
| Distribution learning | QM9 | ~134K molecules | Small organic molecules |
Algorithms
- Fragmentation: JTVAE, BRICS, MMPA, Scaffold (all via RDKit)
- Tree construction: AMT from reduced graph, then FBT transformation
- Traversal: Breadth-first search on FBT
- Generative model: MolGPT (Transformer decoder)
- Discriminative model: AttentiveFP for activity prediction on JNK3/AID1706
Evaluation
| Metric | Description |
|---|---|
| Validity | Fraction of generated strings that decode to valid molecules |
| Uniqueness | Fraction of distinct molecules among valid generations |
| Novelty | Fraction of generated molecules not in training set |
| KLD | Kullback-Leibler divergence for physicochemical property distributions |
| FCD | Frechet ChemNet Distance measuring chemical similarity to training set |
| Active Novel | Novel molecules predicted active by AttentiveFP |
Artifacts
| Artifact | Type | License | Notes |
|---|---|---|---|
| t-SMILES GitHub | Code | MIT | Official implementation with training/generation scripts |
| Zenodo deposit | Code + Data | CC-BY-4.0 | Archived code and data |
| Code Ocean capsule | Code | Not specified | Certified reproducible compute capsule |
Hardware
The paper mentions limited computational resources but does not specify exact GPU types or training times.
Paper Information
Citation: Wu, J.-N., Wang, T., Chen, Y., Tang, L.-J., Wu, H.-L., & Yu, R.-Q. (2024). t-SMILES: a fragment-based molecular representation framework for de novo ligand design. Nature Communications, 15, 4993.
@article{wu2024tsmiles,
title={t-SMILES: a fragment-based molecular representation framework for de novo ligand design},
author={Wu, Juan-Ni and Wang, Tong and Chen, Yue and Tang, Li-Juan and Wu, Hai-Long and Yu, Ru-Qin},
journal={Nature Communications},
volume={15},
number={1},
pages={4993},
year={2024},
doi={10.1038/s41467-024-49388-6}
}