A Method for Complete Chemical Tokenization
This is a Method paper that introduces two new tokenizers for molecular foundation models: Smirk and Smirk-GPE. The primary contribution is a tokenization scheme that achieves complete coverage of the OpenSMILES specification using only 165 tokens, addressing the vocabulary gaps present in existing atom-wise tokenizers. The paper also proposes n-gram language models as low-cost proxy evaluators for tokenizer quality and validates these proxies against 18 transformer-based models across multiple benchmarks.
Vocabulary Gaps in Molecular Tokenization
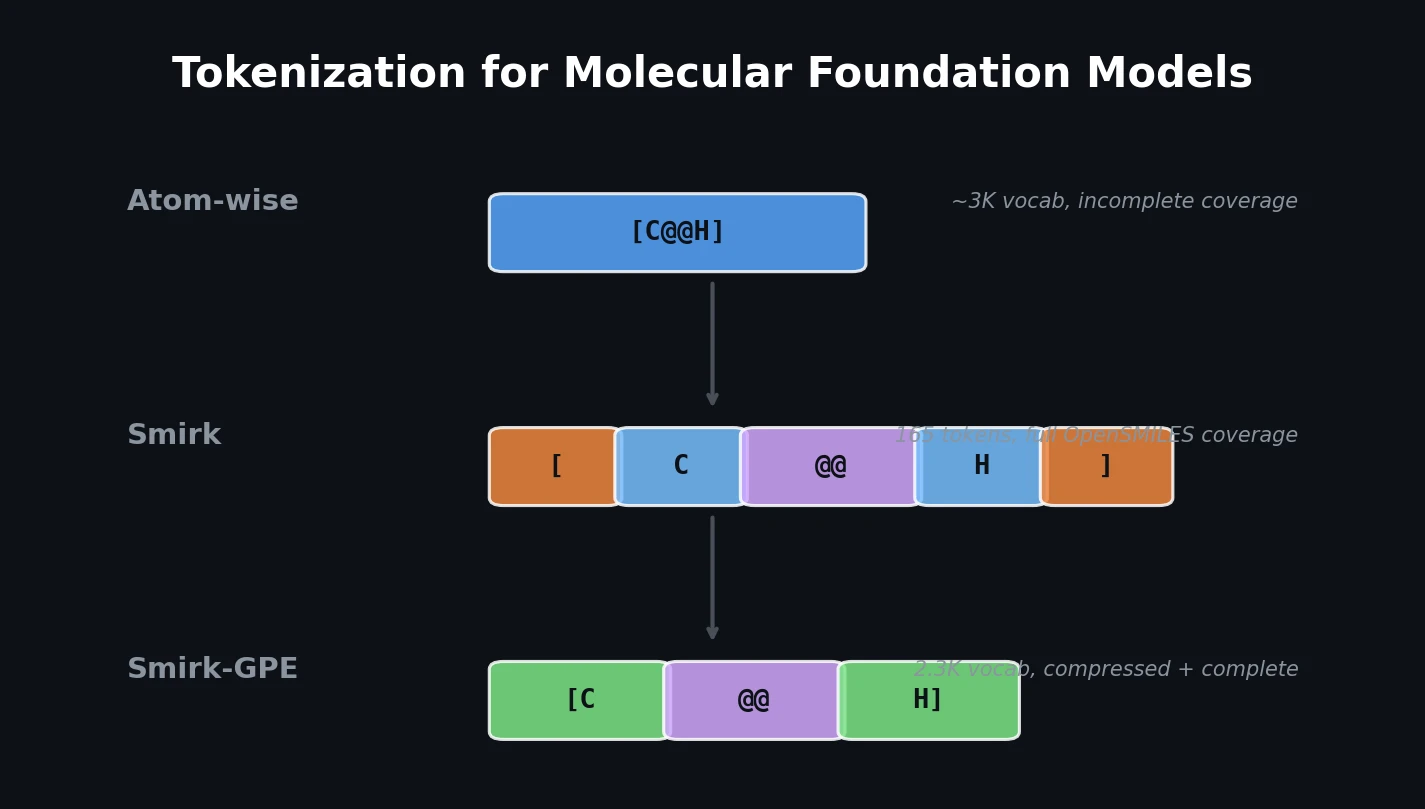
Molecular foundation models overwhelmingly use “atom-wise” tokenization, where SMILES strings are split at atom boundaries using a regular expression first proposed by Schwaller et al. A key pattern in this regex treats all “bracketed atoms” (e.g., [C@@H], [18F], [Au+]) as single, irreducible tokens. Since bracketed atoms encode isotopes, chirality, charge, hydrogen count, and element identity, the number of possible permutations under the OpenSMILES specification exceeds 28 trillion. In practice, existing atom-wise tokenizers maintain vocabularies of fewer than 3,000 tokens, leaving large portions of chemical space unrepresentable.
This gap has real consequences. Many chemistry-specific tokenizers emit the unknown token [UNK] at non-negligible frequencies, particularly on datasets with diverse elements and stereochemistry. For example, SPE and APE tokenizers produce [UNK] for roughly 19% of tokens on MoleculeNet and approximately 50% on the tmQM transition metal complex dataset. Even models like SELFormer and ReactionT5 lack tokens for elements such as copper, ruthenium, gold, and uranium.
The authors also note a subtler issue: some open-vocabulary tokenizers (e.g., ChemBERTa’s BPE) conflate chemically distinct entities. The same Sc token may represent both a sulfur-carbon bond (in organic SMILES) and the element scandium (in [Sc]), creating ambiguity in downstream analysis.
Smirk: Glyph-Level Decomposition of SMILES
The core insight behind Smirk is to fully decompose bracketed atoms into their constituent “glyphs,” the primitive symbols defined by the OpenSMILES specification (element symbols, chirality markers, charges, isotope numbers, hydrogen counts, and brackets themselves). This transforms tokenization from a word-level scheme (one token per bracketed atom) to a character-level scheme over chemically meaningful glyphs.
Smirk uses a two-stage tokenization process:
- Atom decomposition: Split a SMILES string into atom-level units using a regex (e.g.,
OC[C@@H][OH]becomesO C [C@@H] [OH]). - Glyph decomposition: Further split each unit into its constituent glyphs (e.g.,
[C@@H]becomes[ C @@ H ]).
The two-stage process is necessary to resolve ambiguities. For example, Sc in an unbracketed context represents a sulfur-carbon bond, while [Sc] denotes scandium. This ambiguity occurs over half a million times in PubChem’s compound dataset.
The resulting vocabulary contains only 165 tokens, requires no training, and by construction can faithfully tokenize any molecule that conforms to the OpenSMILES specification. The implementation is written in Rust using HuggingFace’s Tokenizers library and is available on PyPI.
Smirk-GPE (Glyph Pair Encoding) extends Smirk with a BPE-like compression step. After Smirk tokenization, adjacent tokens are merged using learned rules, reducing sequence length. Unlike standard BPE, merges operate on token IDs rather than character strings, preserving the distinction between chemically different entities that happen to share the same characters. Smirk-GPE was trained on 262 million molecules from Enamine REAL Space with a target vocabulary of 50,000 tokens, though training terminated at 2,300 tokens after exhausting all possible merges.
Evaluation Framework: Intrinsic Metrics, N-Gram Proxies, and Transformer Benchmarks
The evaluation covers 34 tokenizers across three datasets (Enamine REALSpace, MoleculeNet, and tmQM) using both intrinsic and extrinsic metrics.
Intrinsic Metrics
Four intrinsic metrics are computed for each tokenizer:
Fertility measures the mean tokenized sequence length. Higher fertility increases computational cost due to the quadratic scaling of attention:
$$ \text{cost} \propto \text{fertility}^2 $$
Normalized entropy quantifies how close a tokenizer comes to the information-theoretic ideal where all tokens are equally probable:
$$ \eta = \frac{-1}{\log |V|} \sum_{x \in V} p(x) \log p(x) $$
where $V$ is the vocabulary and $p(x)$ is the observed token probability. Higher normalized entropy correlates with better downstream performance.
Token imbalance measures the distance between observed token frequencies and a uniform distribution:
$$ D = \frac{1}{2} \sum_{x \in V} |p(x) - |V|^{-1}| $$
Unknown token frequency captures the fraction of emitted tokens that are [UNK]. This metric is particularly revealing: all existing chemistry-specific tokenizers (SPE/APE, atom-wise, BPE, and Unigram variants) emit [UNK] at non-negligible rates, while NLP tokenizers, Smirk, and Smirk-GPE do not.
N-Gram Proxy Language Models
The paper proposes using n-gram models as low-cost proxies for transformer-based evaluation. An n-gram estimates token likelihood with add-one smoothing:
$$ P_{n}(x_{i} \mid x_{i-n+1}, \dots, x_{i-1}) = \frac{C(x_{i-n+1}, \dots, x_{i}) + 1}{C(x_{i-n+1}, \dots, x_{i-1}) + |V|} $$
where $C$ is the count function and $|V|$ is the vocabulary size. N-grams were “pretrained” on 1.6 billion SMILES from Enamine REAL Space and evaluated on validation splits. Cross-entropy loss and information loss from unknown tokens were computed.
To quantify information lost to [UNK] tokens, the authors compute the KL-divergence between token distributions with and without unknown tokens, using a bidirectional character n-gram model:
$$ B_{n}(x_{i} \mid x_{i-n+1}, \dots, x_{i-1}, x_{i+1}, \dots, x_{i+n-1}) \propto \frac{C(x_{i-n+1}, \dots, x_{i}) + 1}{C(x_{i-n+1}, \dots, x_{i-1}) + |V|} \times \frac{C(x_{i}, \dots, x_{i+n-1}) + 1}{C(x_{i+1}, \dots, x_{i+n-1}) + |V|} $$
Transformer Experiments
Eighteen encoder-only RoBERTa models (25M parameters each, excluding embeddings) were pretrained from scratch using masked language modeling on Enamine REAL Space (245M molecules, 30,000 steps). Each model used a different tokenizer, isolating the tokenizer’s effect on performance. Finetuning was conducted on six regression and seven classification tasks from MoleculeNet and tmQM.
Linear fixed-effects models were used to estimate the standardized effect of each tokenization scheme relative to an atom-wise SMILES baseline.
Key Findings and Practical Implications
Tokenizer Performance
- Smirk shows a positive effect on pretraining quality and downstream performance on tmQM (the dataset with the most bracketed atoms), but performs comparably to atom-wise tokenization on MoleculeNet tasks.
- SPE and APE tokenizers have a negative impact on both pretraining and downstream performance relative to the atom-wise baseline, likely due to their high
[UNK]rates. - Molecular encoding choice (SMILES vs. SELFIES) has a negligible effect on performance.
- NLP tokenizers (GPT-4o, LLaMA, Gemma) score comparably to chemistry-specific tokenizers on intrinsic metrics and do not emit unknown tokens.
N-Gram Proxy Validation
N-gram cross-entropy and information loss metrics show strong rank correlation (Spearman’s $\rho$) with downstream transformer performance, validating their use as low-cost evaluation proxies. The effect sizes from n-gram and transformer experiments are directionally consistent.
Information Loss from Unknown Tokens
Information loss is minimal for tokenizers with robust coverage but substantial for tokenizers with limited vocabularies on chemically diverse datasets. MoLFormer incurs only 0.1 nats/molecule on MoleculeNet but 40.3 nats/molecule on tmQM. Open-vocabulary tokenizers (Smirk, Smirk-GPE, NLP tokenizers) mitigate this degradation.
Practical Recommendations
The authors argue that molecular foundation models must encode the entire breadth of chemical space or risk obscuring critical features. Bracketed atoms encode information essential to clinically relevant pharmaceuticals (e.g., Amoxicillin), industrial compounds (e.g., Tricalcium Silicate), and foundational chemistry (e.g., Cisplatin, where omitting the chiral marker erases medically relevant stereochemical information). The paper encourages the community to adopt open-vocabulary tokenizers and develop more chemically diverse benchmarks.
Limitations
- The analysis uses a single-point evaluation for transformer experiments, which may underestimate performance achievable with additional hyperparameter tuning.
- Smirk-GPE’s learned merges from REALSpace did not fully generalize to tmQM, as indicated by the token imbalance metric.
- Current benchmarks (MoleculeNet) lack sufficient diversity to evaluate tokenizer robustness across the full periodic table, isotopes, charged species, and uncommon bond types.
- The downstream impact of token ambiguities in BPE-based tokenizers (e.g., ChemBERTa’s conflation of
Scas both sulfur-carbon and scandium) remains unclear.
Reproducibility Details
Data
| Purpose | Dataset | Size | Notes |
|---|---|---|---|
| Pretraining | Enamine REAL Space | 1.6B SMILES (n-gram), 245M molecules (transformer) | 80/10/10 train/val/test split |
| Downstream evaluation | MoleculeNet | Multiple tasks | 6 regression + 7 classification tasks |
| Downstream evaluation | tmQM | 108K transition metal complexes | OpenSMILES molecular encodings |
| Smirk-GPE training | Enamine REAL Space (subset) | 262M molecules | Training split only |
Algorithms
- Smirk: Two-stage regex-based tokenization (atom decomposition, then glyph decomposition). No training required. Vocabulary: 165 tokens.
- Smirk-GPE: BPE-like compression on top of Smirk. Operates on token IDs (not strings) to preserve chemical disambiguation. Final vocabulary: 2,300 tokens.
- N-gram models: Add-one smoothing, bidirectional context ($2n - 2$ total context window). Implemented in Julia with exact integer arithmetic.
Models
- Architecture: RoBERTa-PreLayerNorm, 8 layers, 8 attention heads, hidden size 512, intermediate size 2048, max sequence length 2048. ~25M parameters (excluding embeddings).
- Pretraining: Masked language modeling, 30,000 steps, effective batch size 8192, FusedLamb optimizer, learning rate $1.6 \times 10^{-4}$.
- Finetuning: 100,000 steps, AdamW optimizer, effective batch size 128, learning rate $1.6 \times 10^{-4}$.
Evaluation
- MoleculeNet preferred metrics per task (AUROC for classification, MAE/RMSE for regression)
- Fixed-effects models for standardized effect size estimation
- Spearman’s rank correlation between n-gram and transformer metrics
Hardware
- Pretraining: 2x NVIDIA A100 GPUs (Delta system at NCSA)
- Finetuning: 1x NVIDIA A40 GPU
- N-gram models: CPU-based (Julia implementation)
Artifacts
| Artifact | Type | License | Notes |
|---|---|---|---|
| Smirk tokenizer | Code | Apache-2.0 | Rust implementation with Python bindings, available on PyPI |
| Model checkpoints | Model | Not specified | Pretrained and finetuned checkpoints included in data release |
| N-gram code | Code | Not specified | Julia implementation included in data release |
Paper Information
Citation: Wadell, A., Bhutani, A., & Viswanathan, V. (2026). Tokenization for Molecular Foundation Models. Journal of Chemical Information and Modeling, 66(3), 1384-1393. https://doi.org/10.1021/acs.jcim.5c01856
@article{wadell2026tokenization,
title={Tokenization for Molecular Foundation Models},
author={Wadell, Alexius and Bhutani, Anoushka and Viswanathan, Venkatasubramanian},
journal={Journal of Chemical Information and Modeling},
volume={66},
number={3},
pages={1384--1393},
year={2026},
publisher={American Chemical Society},
doi={10.1021/acs.jcim.5c01856}
}