A Data-Driven Tokenization Method for Chemical Deep Learning
This is a Method paper that introduces SMILES Pair Encoding (SPE), a tokenization algorithm adapted from byte pair encoding (BPE) in natural language processing. The primary contribution is a data-driven approach that learns a vocabulary of high-frequency SMILES substrings from a large chemical dataset and then uses that vocabulary to tokenize SMILES for downstream deep learning tasks. The authors provide an open-source Python package (SmilesPE) and demonstrate improvements on both molecular generation and QSAR prediction benchmarks.
Limitations of Atom-Level SMILES Tokenization
SMILES-based deep learning models require tokenization to convert molecular strings into sequences of discrete units. The standard approaches have well-known drawbacks:
- Character-level tokenization breaks SMILES character by character, splitting chemically meaningful multi-character atoms. For example,
[C@@H]becomes six separate tokens ([,C,@,@,H,]), losing the stereochemistry information of a single carbon. - Atom-level tokenization addresses some of these issues by treating multi-character element symbols (Cl, Br) and bracketed atoms ([nH], [O-]) as single tokens. However, these tokens still encode only individual atoms, not substructures.
- k-mer tokenization (sequences of k consecutive overlapping characters) captures some connectivity information but suffers from the out-of-vocabulary problem: the model cannot represent k-mers not seen during training.
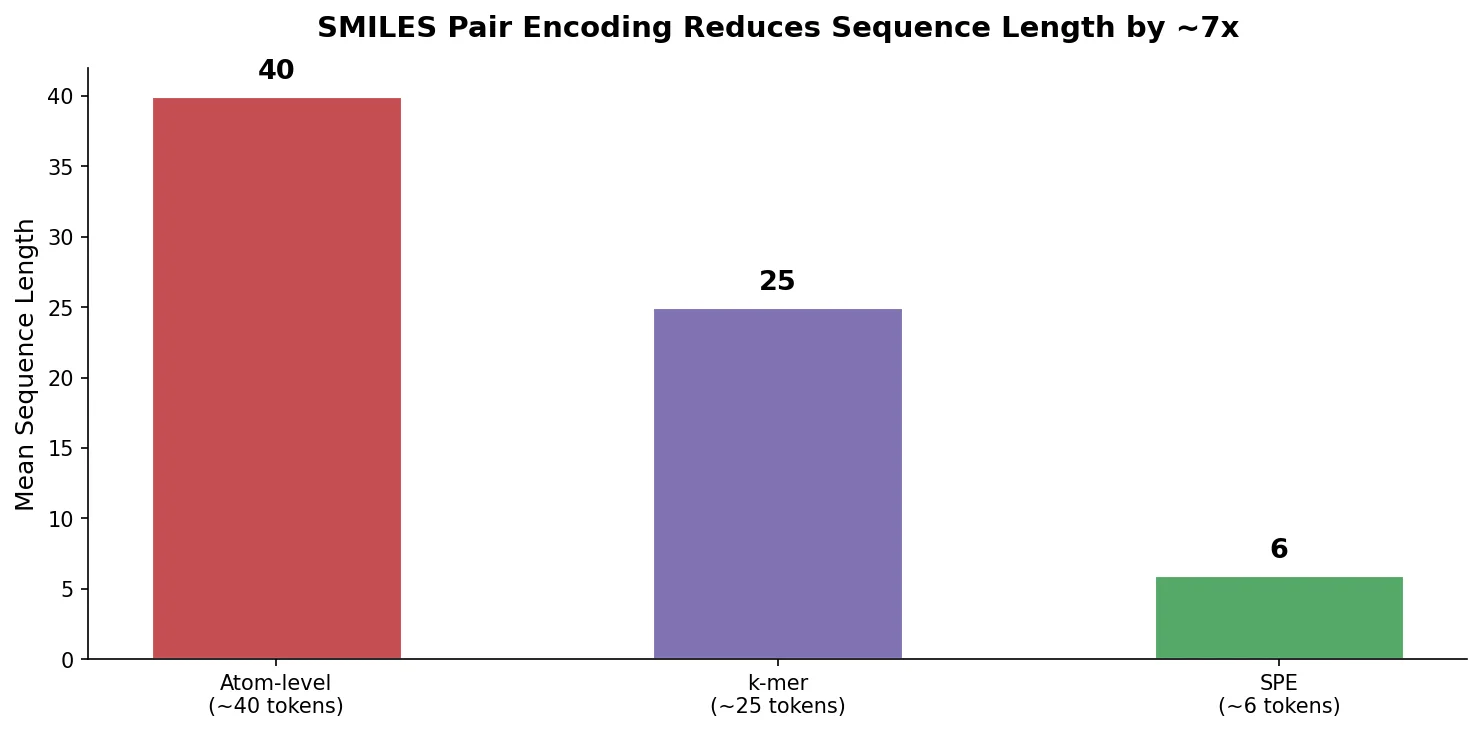
All three approaches produce relatively long input sequences (mean ~40 tokens per molecule on ChEMBL at the atom level), which increases computational cost for sequential architectures like RNNs and exacerbates long-range dependency issues.
Core Innovation: Adapting Byte Pair Encoding for SMILES
SPE adapts the byte pair encoding algorithm, originally developed for data compression and later adopted for subword tokenization in NLP, to the domain of chemical strings. The algorithm has two phases:
Vocabulary training:
- Tokenize SMILES from a large dataset (ChEMBL) at the atom level
- Initialize the vocabulary with all unique atom-level tokens
- Iteratively count the frequency of all adjacent token pairs, merge the most frequent pair into a new token, and add it to the vocabulary
- Stop when either the maximum vocabulary size (MVS) or a minimum frequency threshold (FT) is reached
Tokenization: Given a trained SPE vocabulary, a new SMILES string is first tokenized at the atom level, then token pairs are iteratively merged according to their frequency rank in the vocabulary until no further merges are possible.
The key hyperparameters are MVS and FT. In the reported experiments, MVS was set to 30,000 and FT was set to 2,000. The vocabulary was trained on ~3.4 million SMILES (both canonical and one non-canonical variant per molecule) from ChEMBL25. The resulting vocabulary contained 3,002 unique SMILES substrings with lengths ranging from 1 to 22 atom-level characters.
The trained SPE vocabulary produces tokens that are human-readable and correspond to chemically meaningful substructures and functional groups. SPE tokenization reduces the mean sequence length from approximately 40 tokens (atom-level) to approximately 6 tokens on ChEMBL, a roughly 6-7x compression. This shorter representation directly reduces computational cost for RNN-based and other sequential models.
The algorithm is also compatible with other text-based molecular representations such as DeepSMILES and SELFIES, since these share atom-level character structures that can serve as the starting point for pair merging.
Molecular Generation and QSAR Prediction Experiments
Molecular Generation
The authors trained AWD-LSTM language models with SPE and atom-level tokenization on 9 million SMILES (1 canonical + 5 non-canonical per compound from ChEMBL25). Each model sampled 1 million SMILES for evaluation. The AWD-LSTM architecture used an embedding size of 400, three LSTM layers with 1,152 hidden units each, and various dropout settings (embedding: 0.1, input: 0.6, weight: 0.5, hidden: 0.2). Models were trained for 10 epochs with a base learning rate of 0.008 using one-cycle scheduling.
| Metric | SPE | Atom-level |
|---|---|---|
| Validity | 0.941 | 0.970 |
| Uniqueness | 0.994 | 0.992 |
| Novelty | 0.983 | 0.978 |
| Internal diversity | 0.897 | 0.886 |
| Nearest neighbor similarity | 0.391 | 0.386 |
The SPE model generated a more diverse population of novel molecules at the cost of slightly lower validity (94.1% vs. 97.0%). Internal diversity is defined as:
$$ \text{Internal diversity} = 1 - \frac{1}{|G|} \sum_{(x_1, x_2) \in G \times G} T(x_1, x_2) $$
where $T(x_1, x_2)$ is the Tanimoto similarity between molecules $x_1$ and $x_2$ using 1024-bit ECFP6 fingerprints. Nearest neighbor similarity (SNN) measures how well the generated set resembles the reference set:
$$ \text{SNN} = \frac{1}{|G|} \sum_{x_G \in G} \max_{x_R \in R} T(x_G, x_R) $$
Substructure coverage analysis showed both models recovered the same top-1000 BRICS fragments (100% coverage), but SPE consistently outperformed atom-level tokenization on top-5000 coverage across all four substructure types: BRICS fragments (0.997 vs. 0.987), functional groups (0.688 vs. 0.659), scaffolds (0.872 vs. 0.825), and ring systems (0.781 vs. 0.761).
QSAR Prediction
QSAR models were built using the MolPMoFiT transfer learning framework, which pre-trains a language model on ChEMBL and then fine-tunes it for specific prediction tasks. The evaluation used 24 regression benchmarks (pIC50 values) from Cortes-Ciriano et al., covering targets ranging from 199 molecules (alpha-2a adrenergic receptor) to 5,010 molecules (hERG). Models were evaluated on 10 random 80:10:10 splits using RMSE, R-squared, and MAE. Random forest models with 1024-bit ECFP6 were included as baseline comparisons.
Cohen’s d effect sizes were computed to quantify performance differences between tokenization methods. SPE performed comparably or better than atom-level tokenization on 23 out of 24 datasets. Notable results with medium or large effect sizes favoring SPE included cannabinoid CB1 receptor (large effect), A2a adrenergic receptor, LCK, estrogen receptor, and Aurora-A kinase (all medium effects). Against k-mer tokenization, SPE matched or outperformed on 22 out of 24 datasets.
Cohen’s d is defined as:
$$ \text{Cohen’s } d = \frac{\bar{x}_1 - \bar{x}_2}{\sqrt{(\text{SD}_1^2 + \text{SD}_2^2) / 2}} $$
where $\bar{x}_1, \bar{x}_2$ are the group means and $\text{SD}_1, \text{SD}_2$ are the standard deviations. Thresholds of 0.2 (small), 0.5 (medium), and 0.8 (large) were used following standard recommendations.
SMILES-based deep learning models generally performed on par with or better than the RF baseline, with particularly strong advantages on the four largest datasets (COX-2, acetylcholinesterase, erbB1, and hERG).
In addition to performance gains, SPE-based models trained on average 5 times faster than atom-level models due to the shorter input sequences.
Results Summary and Future Directions
The main findings of this study are:
SPE produces chemically meaningful tokens. The learned vocabulary contains human-readable SMILES substrings that correspond to common substructures and functional groups, making model interpretations more accessible.
SPE compresses input sequences by ~6-7x. Mean token sequence length drops from ~40 (atom-level) to ~6 (SPE) on ChEMBL, yielding a ~5x training speedup.
SPE improves molecular generation diversity. The SPE-based generative model produces molecules with higher novelty (98.3% vs. 97.8%), internal diversity (0.897 vs. 0.886), and substructure coverage, at the cost of slightly lower validity (94.1% vs. 97.0%).
SPE matches or outperforms atom-level and k-mer tokenization on QSAR prediction. Across 24 benchmarks, SPE showed comparable or better performance in 23/24 comparisons against atom-level and 22/24 against k-mer tokenization.
Limitations acknowledged by the authors:
- The SPE vocabulary is trained on a specific dataset (ChEMBL25) and may not optimally represent chemical spaces that differ significantly from drug-like compounds.
- The validity rate for molecular generation is slightly lower than atom-level tokenization (94.1% vs. 97.0%), since longer substructure tokens can introduce invalid fragments.
- The k-mer tokenization suffers from an out-of-vocabulary problem, which the authors address by replacing unseen 4-mers with
[UNK]tokens, but this is a limitation of the comparison rather than of SPE itself.
Future directions: The authors suggest SPE could serve as a general tokenization method for SMILES-based deep learning, applicable to any task where SMILES strings are used as input (generation, property prediction, reaction prediction, retrosynthesis). The algorithm can also be applied to DeepSMILES and SELFIES representations without modification.
Reproducibility Details
Data
| Purpose | Dataset | Size | Notes |
|---|---|---|---|
| SPE vocabulary training | ChEMBL25 | ~3.4M SMILES | 1 canonical + 1 non-canonical per molecule |
| Language model training | ChEMBL25 augmented | ~9M SMILES | 1 canonical + 5 non-canonical per molecule |
| Molecular generation evaluation | Sampled from model | 1M SMILES per model | Validated with RDKit |
| QSAR benchmarks | Cortes-Ciriano et al. | 24 datasets, 199-5010 molecules | pIC50 regression tasks |
Algorithms
- SPE vocabulary training: iterative pair merging with MVS=30,000 and FT=2,000
- Language model: AWD-LSTM with embedding size 400, 3 LSTM layers with 1,152 hidden units
- Dropout: embedding=0.1, input=0.6, weight=0.5, hidden=0.2
- Training: 10 epochs, base learning rate 0.008, one-cycle policy
- QSAR: MolPMoFiT transfer learning with 25x training augmentation and 15x validation augmentation
- Test time augmentation: average of canonical + 4 augmented SMILES predictions
- RF baseline: 500 trees, 1024-bit ECFP6, default scikit-learn parameters
Models
- AWD-LSTM architecture from Merity et al. (2018)
- MolPMoFiT framework from Li and Fourches (2020) for transfer learning QSAR
Evaluation
| Metric | Task | Notes |
|---|---|---|
| Validity, Uniqueness, Novelty | Generation | Basic quality metrics |
| Internal diversity | Generation | 1 - mean pairwise Tanimoto (ECFP6) |
| Nearest neighbor similarity | Generation | Mean max Tanimoto to reference set |
| Substructure coverage | Generation | BRICS, functional groups, scaffolds, ring systems |
| RMSE, R-squared, MAE | QSAR regression | 10 random 80:10:10 splits |
| Cohen’s d | QSAR comparison | Effect size between tokenization methods |
Hardware
Not explicitly specified in the paper.
Artifacts
| Artifact | Type | License | Notes |
|---|---|---|---|
| SmilesPE | Code | Apache-2.0 | SPE tokenization Python package |
| MolPMoFiT | Code | Not specified | Transfer learning QSAR framework |
Paper Information
Citation: Li, X., & Fourches, D. (2021). SMILES Pair Encoding: A Data-Driven Substructure Tokenization Algorithm for Deep Learning. Journal of Chemical Information and Modeling, 61(4), 1560-1569. https://doi.org/10.1021/acs.jcim.0c01127
@article{li2021smiles,
title={SMILES Pair Encoding: A Data-Driven Substructure Tokenization Algorithm for Deep Learning},
author={Li, Xinhao and Fourches, Denis},
journal={Journal of Chemical Information and Modeling},
volume={61},
number={4},
pages={1560--1569},
year={2021},
publisher={American Chemical Society},
doi={10.1021/acs.jcim.0c01127}
}