Data Augmentation Through SMILES Randomization
This is an Empirical paper that performs an extensive benchmark of RNN-based molecular generative models trained with different SMILES string variants. The primary contribution is demonstrating that randomized SMILES (non-unique molecular string representations obtained by randomizing atom orderings) substantially improve the quality of the generated chemical space compared to canonical SMILES, without requiring any changes to the model architecture.
The paper evaluates three properties of generated chemical spaces: uniformity (equal probability of sampling each molecule), completeness (coverage of the target space), and closedness (generating only molecules within the target space). These are measured using a new composite metric called UC-JSD.
Canonical SMILES Bias in Generative Models
Recurrent Neural Networks trained on SMILES strings have shown the capacity to create large chemical spaces of valid molecules. However, when trained with canonical SMILES (the unique string representation produced by a canonicalization algorithm), these models exhibit biases. Specifically, prior work by the same group showed that models trained on one million GDB-13 molecules could only recover 68% of GDB-13 when sampled two billion times, compared to the theoretical maximum of 87% from an ideal uniform sampler.
The canonical SMILES representation introduces two problems. First, the canonicalization algorithm constrains how the molecular graph is traversed (e.g., prioritizing sidechains over ring atoms), forcing the model to learn both valid SMILES syntax and the specific canonical ordering rules. Second, structurally similar molecules can have substantially different canonical SMILES, making some molecules harder to sample than others. Molecules with more ring systems and complex topologies are particularly underrepresented.
The authors also note that DeepSMILES, a recently proposed alternative syntax, had not been benchmarked against randomized SMILES, and that the data augmentation capabilities of randomized SMILES at different training set sizes were unexplored.
Randomized SMILES as Non-Canonical Representations
The core insight is that by randomizing the atom ordering before SMILES generation, each molecule can be represented by multiple different but equally valid SMILES strings. This effectively provides data augmentation: a molecule with $n$ heavy atoms can theoretically yield up to $n$ different SMILES strings (though the actual number is typically lower due to molecular symmetry).
Two randomized SMILES variants are explored:
- Restricted randomized SMILES: Atom ordering is randomized, but RDKit’s built-in fixes are applied. These fixes prevent overly complicated traversals, such as prioritizing sidechains before completing ring atoms.
- Unrestricted randomized SMILES: Atom ordering is randomized without any RDKit restrictions, producing a superset of the restricted variant that includes more convoluted SMILES strings.
For each training epoch, a new set of randomized SMILES is generated for the same molecules, so a model trained for 300 epochs on one million molecules sees approximately 300 million different SMILES strings (with some overlap due to sampling).
The model architecture is a standard RNN with an embedding layer, $l$ layers of LSTM or GRU cells of size $w$, optional dropout, and a linear output layer with softmax. The training objective minimizes the average negative log-likelihood (NLL):
$$ J(T) = -\ln P(X_{0} = x_{0}) - \sum_{t=1}^{T} \ln P(X_{t} = x_{t} \mid X_{t-1} = x_{t-1} \dots X_{1} = x_{1}) $$
The key metric is the Uniformity-Completeness JSD (UC-JSD), which extends the Jensen-Shannon Divergence to measure how uniform, complete, and closed the generated chemical space is:
$$ JSD = H\left(\sum_{d \in D} \alpha_{i} \cdot d_{i}\right) - \sum_{d \in D} \alpha_{i} H(d_{i}) $$
where $H(d)$ is the Shannon entropy of a probability distribution. The UC-JSD is computed over the NLL vectors of the validation, training, and sampled sets. The composite UCC score is defined as:
$$ UCC = \text{completeness} \times \text{uniformity} \times \text{closedness} $$
where completeness measures coverage of GDB-13, uniformity measures how equal the sampling probabilities are, and closedness measures how few invalid (out-of-target-space) molecules are generated.
Benchmark Design Across SMILES Variants, Training Sizes, and Architectures
The benchmark covers a systematic grid of experimental conditions:
SMILES variants: Canonical, restricted randomized, unrestricted randomized, and three DeepSMILES variants (branch syntax, ring syntax, both).
Training set sizes from GDB-13: 1,000,000, 10,000, and 1,000 molecules with corresponding validation sets.
Architecture choices: LSTM vs. GRU cells, with hyperparameter grids over number of layers ($l$), hidden size ($w$), dropout rate ($d$), and batch size ($b$).
| Model | Layers ($l$) | Hidden ($w$) | Dropout ($d$) | Batch ($b$) | Cell |
|---|---|---|---|---|---|
| GDB-13 1M | 3 | 512 | 0, 25, 50 | 64, 128, 256, 512 | GRU, LSTM |
| GDB-13 10K | 2, 3, 4 | 256, 384, 512 | 0, 25, 50 | 8, 16, 32 | LSTM |
| GDB-13 1K | 2, 3, 4 | 128, 192, 256 | 0, 25, 50 | 4, 8, 16 | LSTM |
| ChEMBL | 3 | 512 | 0, 25, 50 | 64, 128, 256, 512 | LSTM |
Each model’s best epoch was selected using a smoothed UC-JSD curve, and the best epoch was then sampled with replacement $k = 2 \times 10^{9}$ times for GDB-13 benchmarks.
For ChEMBL experiments, models were trained on 1,483,943 molecules with a validation set of 78,102 molecules. Evaluation used validity, unique molecule count, and Frechet ChemNet Distance (FCD).
Randomized SMILES Produce More Complete and Uniform Chemical Spaces
GDB-13 results (1M training set)
The restricted randomized SMILES model recovered 83.0% of GDB-13, compared to 72.8% for canonical SMILES and 68.4-72.1% for DeepSMILES variants. All three quality metrics improved substantially:
| SMILES Variant | % GDB-13 | Uniformity | Completeness | Closedness | UCC |
|---|---|---|---|---|---|
| Canonical | 72.8 | 0.879 | 0.836 | 0.861 | 0.633 |
| Rand. restricted | 83.0 | 0.977 | 0.953 | 0.925 | 0.860 |
| Rand. unrestricted | 80.9 | 0.970 | 0.929 | 0.876 | 0.790 |
| DeepSMILES (both) | 68.4 | 0.851 | 0.785 | 0.796 | 0.532 |
The NLL distribution of GDB-13 molecules under the randomized SMILES model was centered near $NLL_{GDB13} = -\ln(1/|GDB13|) = 20.6$ with a narrow spread, indicating near-uniform sampling probability. The canonical model showed a much wider NLL distribution, meaning some molecules were orders of magnitude harder to sample.
Randomized SMILES without data augmentation (same SMILES each epoch) still outperformed canonical SMILES (UCC 0.712 vs. 0.633 for restricted), confirming that the non-canonical representation itself is beneficial beyond the augmentation effect.
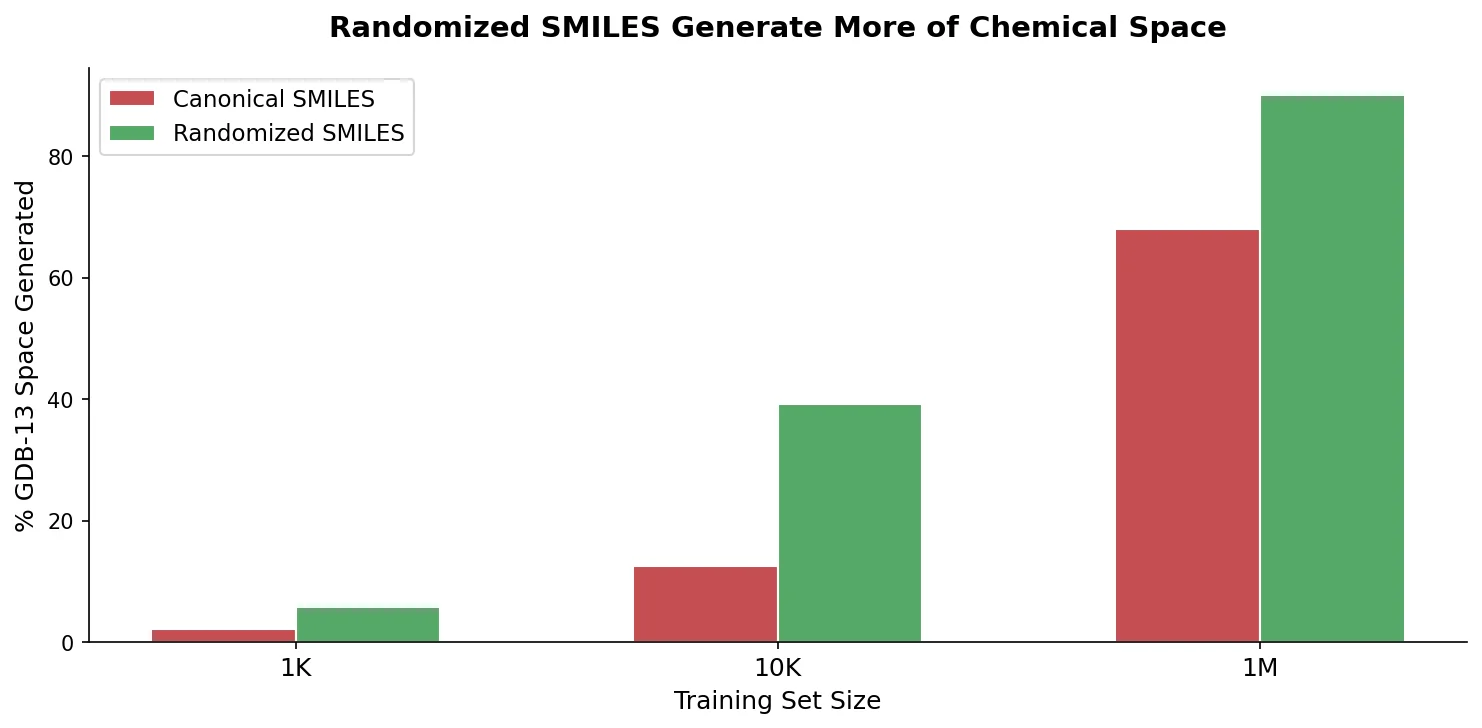
Smaller training sets amplify the advantage
With only 10,000 training molecules (0.001% of GDB-13), the randomized model generated 62.3% of GDB-13 vs. 38.8% for canonical. With 1,000 training molecules, the gap widened further: 34.1% vs. 14.5%. Validity also improved dramatically (81.2% vs. 50.4% for the 1K setting), suggesting randomized SMILES helps the model learn valid SMILES syntax more effectively from limited data.
ChEMBL results
On the drug-like ChEMBL dataset, the randomized SMILES model generated at least double the number of unique molecules compared to canonical (64.09% vs. 34.67% unique in a 2B sample), with comparable validity (98.33% vs. 98.26%). The canonical model showed a lower FCD (0.0712 vs. 0.1265), but the authors argue this reflects overfitting: the canonical model’s NLL distributions for training and validation sets overlapped tightly, while the randomized model showed more uniform coverage. Physicochemical property distributions (molecular weight, logP, SA score, QED, NP score, internal diversity) were nearly identical across both models.
Architecture findings
LSTM cells consistently outperformed GRU cells across all SMILES variants. Despite GRU’s faster per-epoch training time, LSTM models converged in fewer epochs, making them faster overall. Dropout improved canonical SMILES models but was less beneficial (or detrimental) for randomized SMILES, suggesting that randomized SMILES themselves serve as a regularization mechanism. Larger batch sizes generally improved performance across all variants.
UC-JSD as a model selection metric
The UC-JSD showed strong correlation with UCC ($R^{2} = 0.931$ for canonical, $R^{2} = 0.856$ for restricted randomized, $R^{2} = 0.885$ for unrestricted randomized), validating its use as a model selection criterion without requiring expensive sampling of every model.
The authors interpret randomized SMILES models as occupying a hybrid space between grammar-based and action-based generative models. The vocabulary serves as a fixed action space where atom tokens are “add atom” actions, bond tokens are “add bond” actions, and ring/branching tokens enable graph traversal. Canonical SMILES constrain this action space to a single deterministic path, while randomized SMILES allow the model to explore multiple valid traversals. This perspective also explains why DeepSMILES performed worse: its altered syntax creates a more complex action space without compensating benefits.
The authors encourage the use of randomized SMILES across different model architectures and tasks, including classification and property prediction, and suggest that finding optimal restricted variants of randomized SMILES is a promising research direction.
Reproducibility Details
Data
| Purpose | Dataset | Size | Notes |
|---|---|---|---|
| Training/Eval | GDB-13 subsets | 1M / 10K / 1K molecules | Randomly sampled from 975M GDB-13 |
| Training/Eval | ChEMBL | 1,483,943 training / 78,102 validation | Filtered subset of ChEMBL database |
GDB-13 is available from the Reymond group website. ChEMBL is publicly available.
Algorithms
- Character-level tokenization with special handling for multi-character tokens (Cl, Br, bracketed atoms, %-prefixed ring numbers)
- Teacher forcing during training with NLL loss
- Gradient norm clipping to 1.0
- Weight initialization from $\mathcal{U}(-\sqrt{1/w}, \sqrt{1/w})$
- Adaptive learning rate decay based on UC-JSD
- Best epoch selection via smoothed UC-JSD (window size 4)
Models
Standard RNN architecture: embedding layer, stacked LSTM/GRU layers with optional dropout, linear output with softmax. Best models used 3 layers of 512-dimensional LSTM cells. Vocabulary sizes: 26 (GDB-13), 31 (ChEMBL).
Evaluation
| Metric | Best Randomized | Best Canonical | Notes |
|---|---|---|---|
| % GDB-13 (1M) | 83.0% | 72.8% | 2B sample with replacement |
| UCC (1M) | 0.860 | 0.633 | Composite score |
| % GDB-13 (10K) | 62.3% | 38.8% | 2B sample with replacement |
| % GDB-13 (1K) | 34.1% | 14.5% | 2B sample with replacement |
| % Unique ChEMBL | 64.09% | 34.67% | 2B sample with replacement |
Hardware
Nvidia Tesla V100 (Volta) 16 GB VRAM with CUDA 9.1, driver 390.30. Training times ranged from 1 minute (1K canonical) to 131 hours (ChEMBL canonical). Randomized SMILES models required longer per-epoch training due to augmentation overhead but converged to better solutions.
Artifacts
| Artifact | Type | License | Notes |
|---|---|---|---|
| reinvent-randomized | Code | MIT | Training and benchmarking code |
| GDB-13 | Dataset | Academic use | 975 million fragment-like molecules |
| MOSES benchmark | Code | MIT | Used for FCD and property calculations |
Paper Information
Citation: Arús-Pous, J., Johansson, S. V., Prykhodko, O., Bjerrum, E. J., Tyrchan, C., Reymond, J.-L., Chen, H., & Engkvist, O. (2019). Randomized SMILES strings improve the quality of molecular generative models. Journal of Cheminformatics, 11(1), 71. https://doi.org/10.1186/s13321-019-0393-0
@article{aruspous2019randomized,
title={Randomized SMILES strings improve the quality of molecular generative models},
author={Ar{\'u}s-Pous, Josep and Johansson, Simon Viet and Prykhodko, Oleksii and Bjerrum, Esben Jannik and Tyrchan, Christian and Reymond, Jean-Louis and Chen, Hongming and Engkvist, Ola},
journal={Journal of Cheminformatics},
volume={11},
number={1},
pages={71},
year={2019},
doi={10.1186/s13321-019-0393-0},
publisher={Springer}
}