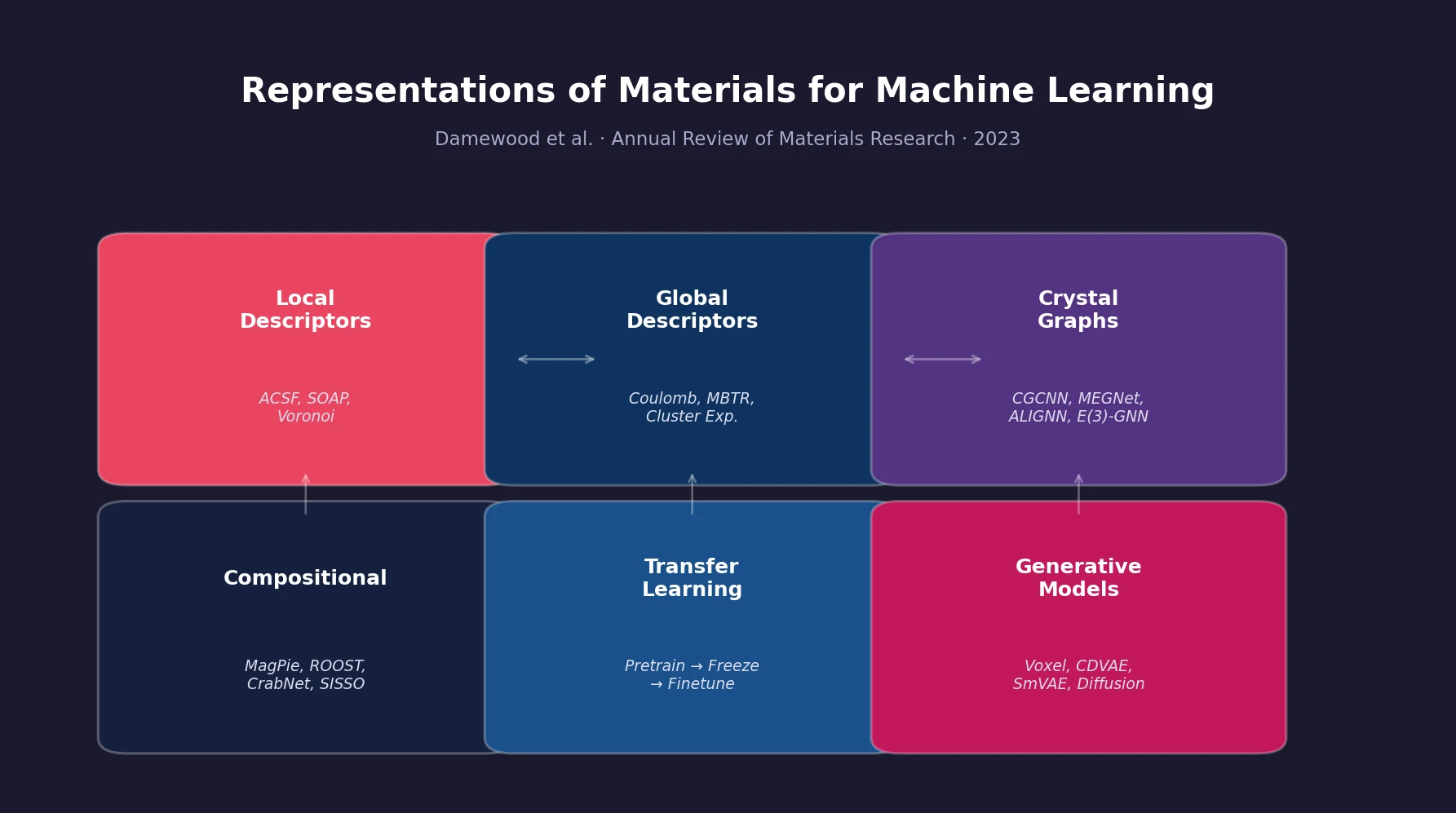
A Systematization of Material Representations
This paper is a Systematization that organizes and categorizes the strategies researchers use to convert solid-state materials into numerical representations suitable for machine learning models. Rather than proposing a new method, the review provides a structured taxonomy of existing approaches, connecting each to the practical constraints of data availability, computational cost, and prediction targets. It covers structural descriptors, graph-based learned representations, compositional features, transfer learning, and generative models for inverse design.
Why Material Representations Matter
Machine learning has enabled rapid property prediction for materials, but every ML pipeline depends on how the material is encoded as a numerical input. The authors identify three guiding principles for effective representations:
- Similarity preservation: Similar materials should have similar representations, and dissimilar materials should diverge in representation space.
- Domain coverage: The representation should be constructable for every material in the target domain.
- Cost efficiency: Computing the representation should be cheaper than computing the target property directly (e.g., via DFT).
In practice, materials scientists face several barriers. Atomistic structures span diverse space groups, supercell sizes, and disorder parameters. Real material performance depends on defects, microstructure, and interfaces. Structural information often requires expensive experimental or computational effort to obtain. Datasets in materials science tend to be small, sparse, and biased toward well-studied systems.
Structural Descriptors: Local, Global, and Topological
The review covers three families of hand-crafted structural descriptors that encode atomic positions and types.
Local Descriptors
Local descriptors characterize the environment around each atom. Atom-centered symmetry functions (ACSF), introduced by Behler and Parrinello, define radial and angular functions:
$$ G_{i}^{1} = \sum_{j \neq i}^{\text{neighbors}} e^{-\eta(R_{ij} - R_{s})^{2}} f_{c}(R_{ij}) $$
$$ G_{i}^{2} = 2^{1-\zeta} \sum_{j,k \neq i}^{\text{neighbors}} (1 + \lambda \cos \theta_{ijk})^{\zeta} e^{-\eta(R_{ij}^{2} + R_{ik}^{2} + R_{jk}^{2})} f_{c}(R_{ij}) f_{c}(R_{ik}) f_{c}(R_{jk}) $$
The Smooth Overlap of Atomic Positions (SOAP), proposed by Bartók et al., defines atomic neighborhood density as a sum of Gaussians and computes a rotationally invariant kernel through expansion in radial functions and spherical harmonics:
$$ \rho_{i}(\mathbf{r}) = \sum_{j} \exp\left(-\frac{|\mathbf{r} - \mathbf{r}_{ij}|^{2}}{2\sigma^{2}}\right) = \sum_{nlm} c_{nlm} g_{n}(\mathbf{r}) Y_{lm}(\hat{\mathbf{r}}) $$
The power spectrum $\mathbf{p}(\mathbf{r}) \equiv \sum_{m} c_{nlm}(c_{n’lm})^{*}$ serves as a vector descriptor of the local environment. SOAP has seen wide adoption both as a similarity metric and as input to ML models.
Voronoi tessellation provides another local approach, segmenting space into cells and extracting features like effective coordination numbers, cell volumes, and neighbor properties.
Global Descriptors
Global descriptors encode the full structure. The Coulomb matrix models electrostatic interactions between atoms:
$$ M_{i,j} = \begin{cases} Z_{i}^{2.4} & \text{for } i = j \\ \frac{Z_{i}Z_{j}}{|r_{i} - r_{j}|} & \text{for } i \neq j \end{cases} $$
Other global methods include partial radial distribution functions (PRDF), the many-body tensor representation (MBTR), and cluster expansions. The Atomic Cluster Expansion (ACE) framework generalizes cluster expansions to continuous environments and has become a foundation for modern deep learning potentials.
Topological Descriptors
Persistent homology from topological data analysis (TDA) identifies geometric features at multiple length scales. Topological descriptors capture pore geometries in porous materials and have outperformed traditional structural descriptors for predicting CO$_{2}$ adsorption in metal-organic frameworks and methane storage in zeolites. A caveat is the $O(N^{3})$ worst-case computational cost per filtration.
Crystal Graph Neural Networks
Graph neural networks bypass manual feature engineering by learning representations directly from structural data. Materials are converted to graphs $G(V, E)$ where nodes represent atoms and edges connect neighbors within a cutoff radius, with periodic boundary conditions.
Key architectures discussed include:
| Model | Key Innovation |
|---|---|
| CGCNN | Crystal graph convolutions for broad property prediction |
| MEGNet | Materials graph networks with global state attributes |
| ALIGNN | Line graph neural networks incorporating three-body angular features |
| Equivariant GNNs | E(3)-equivariant message passing for tensorial properties |
The review identifies several limitations. Graph convolutions based on local neighborhoods can fail to capture long-range interactions or periodicity-dependent properties (e.g., lattice parameters, phonon spectra). Strategies to address this include concatenation with hand-tuned descriptors, plane-wave periodic basis modulation, and reciprocal-space features.
A major practical restriction is the requirement for relaxed atomic positions. Graphs built from unrelaxed crystal prototypes lose information about geometric distortions, degrading accuracy. Approaches to mitigate this include data augmentation with perturbed structures, Bayesian optimization of prototypes, and surrogate force-field relaxation.
Equivariant models that introduce higher-order tensors to node and edge features, constrained to transform correctly under E(3) operations, achieve state-of-the-art accuracy and can match structural descriptor performance even in low-data (~100 datapoints) regimes.
Compositional Descriptors Without Structure
When crystal structures are unavailable, representations can be built purely from stoichiometry and tabulated atomic properties (radii, electronegativity, valence electrons). Despite their simplicity, these methods have distinct advantages: zero computational overhead, accessibility to non-experts, and robustness for high-throughput screening.
Key methods include:
- MagPie: 145 input features derived from elemental properties
- SISSO: Compressive sensing over algebraic combinations of atomic properties, capable of discovering interpretable descriptors (e.g., a new tolerance factor $\tau$ for perovskite stability)
- ElemNet: Deep neural network using only fractional stoichiometry as input, outperforming MagPie with >3,000 training points
- ROOST: Fully-connected compositional graph with attention-based message passing, achieving strong performance with only hundreds of examples
- CrabNet: Self-attention on element embeddings with fractional encoding, handling dopant-level concentrations via log-scale inputs
Compositional models cannot distinguish polymorphs and generally underperform structural approaches. They are most valuable when atomistic resolution is unavailable.
Defects, Surfaces, and Grain Boundaries
The review extends beyond idealized unit cells to practical materials challenges:
Point defects: Representations of the pristine bulk can predict vacancy formation energies through linear relationships with band structure descriptors. Frey et al. proposed using relative differences between defect and parent structure properties, requiring no DFT on the defect itself.
Surfaces and catalysis: Binding energy prediction for catalysis requires representations beyond the bulk unit cell. The d-band center for metals and oxygen 2p-band center for metal oxides serve as simple electronic descriptors, following the Sabatier principle that optimal catalytic activity requires intermediate binding strength. Graph neural networks trained on the Open Catalyst 2020 dataset (>1 million DFT energies) have enabled broader screening, though errors remain high for certain adsorbates and non-metallic surfaces.
Grain boundaries: SOAP descriptors computed for atoms near grain boundaries and clustered into local environment classes can predict grain boundary energy, mobility, and shear coupling. This approach provides interpretable structure-property relationships.
Transfer Learning Across Representations
When target datasets are small, transfer learning leverages representations learned from large, related datasets. The standard procedure involves: (1) pretraining on a large dataset (e.g., all Materials Project formation energies), (2) freezing parameters up to a chosen depth, and (3) either fine-tuning remaining layers or extracting features for a separate model.
Key findings from the review:
- Transfer learning is most effective when the source dataset is orders of magnitude larger than the target
- Physically related tasks transfer better (e.g., Open Catalyst absorption energies transfer well to new adsorbates, less so to unrelated small molecules)
- Earlier neural network layers learn more general representations and transfer better across properties
- Multi-depth feature extraction, combining activations from multiple layers, can improve transfer
- Predictions from surrogate models can serve as additional descriptors, expanding screening domains by orders of magnitude
Generative Models for Crystal Inverse Design
Generative models for solid-state materials face challenges beyond molecular generation: more diverse atomic species, the need to specify both positions and lattice parameters, non-unique definitions (rotations, translations, supercell scaling), and large unit cells (>100 atoms for zeolites and MOFs).
The review traces the progression of approaches:
- Voxel representations: Discretize unit cells into volume elements. Early work (iMatGen, Court et al.) demonstrated feasibility but was restricted to specific chemistries or cubic systems.
- Continuous coordinate models: Point cloud and invertible representations allowed broader chemical spaces but lacked symmetry invariances.
- Symmetry-aware models: Crystal Diffusion VAE (CDVAE) uses periodic graphs and SE(3)-equivariant message passing for translationally and rotationally invariant generation, establishing benchmark tasks for the field.
- Constrained models for porous materials: Approaches like SmVAE represent MOFs through their topological building blocks (RFcodes), ensuring all generated structures are physically valid.
Open Problems and Future Directions
The review highlights four high-impact open questions:
- Local vs. global descriptor trade-offs: Local descriptors (SOAP) excel for short-range interactions but struggle with long-range physics. Global descriptors model periodicity but lack generality across space groups. Combining local and long-range features could provide more universal models.
- Prediction from unrelaxed prototypes: ML force fields can relax structures at a fraction of DFT cost, potentially expanding screening domains. Key questions remain about required training data scale and generalizability.
- Applicability of compositional descriptors: The performance gap between compositional and structural models may be property-dependent, being smaller for properties like band gap that depend on global features rather than local site energies.
- Extensions of generative models: Diffusion-based architectures have improved on voxel approaches for small unit cells, but extending to microstructure, dimensionality, and surface generation remains open.
Reproducibility Details
This paper is a review and does not present new experimental results or release any novel code, data, or models. The paper is open-access (hybrid OA at Annual Reviews) and the arXiv preprint is freely available. The following artifacts table covers key publicly available resources discussed in the review.
Artifacts
| Artifact | Type | License | Notes |
|---|---|---|---|
| arXiv preprint (2301.08813) | Other | arXiv (open access) | Free preprint version |
| Materials Project | Dataset | CC-BY-4.0 | DFT energies, band gaps, structures for >100,000 compounds |
| OQMD | Dataset | CC-BY-4.0 | Open Quantum Materials Database, >600,000 DFT entries |
| Open Catalyst 2020 (OC20) | Dataset | CC-BY-4.0 | >1,000,000 DFT surface adsorption energies |
| AFLOW | Dataset | Public | High-throughput ab initio library, >3,000,000 entries |
| Matminer | Code | BSD | Open-source toolkit for materials data mining and featurization |
Algorithms
The review covers: ACSF, SOAP, Voronoi tessellation, Coulomb matrices, PRDF, MBTR, cluster expansions, ACE, persistent homology, CGCNN, MEGNet, ALIGNN, E(3)-equivariant GNNs, MagPie, SISSO, ElemNet, ROOST, CrabNet, VAE, GAN, and diffusion-based crystal generators.
Hardware
No new experiments are conducted. Hardware requirements vary by the referenced methods (DFT calculations require HPC; GNN training typically requires 1-8 GPUs).
Reproducibility Status
Partially Reproducible: The review paper itself is open-access. All major datasets discussed (Materials Project, OQMD, OC20, AFLOW) are publicly available under permissive licenses. Most referenced model implementations (CGCNN, MEGNet, ALIGNN, ROOST, CDVAE) have open-source code. No novel artifacts are released by the authors.
Paper Information
Citation: Damewood, J., Karaguesian, J., Lunger, J. R., Tan, A. R., Xie, M., Peng, J., & Gómez-Bombarelli, R. (2023). Representations of Materials for Machine Learning. Annual Review of Materials Research, 53. https://doi.org/10.1146/annurev-matsci-080921-085947
Publication: Annual Review of Materials Research, 2023
@article{damewood2023representations,
title={Representations of Materials for Machine Learning},
author={Damewood, James and Karaguesian, Jessica and Lunger, Jaclyn R. and Tan, Aik Rui and Xie, Mingrou and Peng, Jiayu and G{\'o}mez-Bombarelli, Rafael},
journal={Annual Review of Materials Research},
volume={53},
year={2023},
doi={10.1146/annurev-matsci-080921-085947}
}