Paper Contribution: A Systematized Tautomer Database Resource
This is a Resource paper with strong Systematization elements. It provides a comprehensive catalog of 86 tautomeric transformation rules (20 pre-existing CACTVS defaults plus 66 new rules derived from experimental literature), designed to serve as a foundational resource for chemical database systems and the InChI V2 identifier standard. The systematic validation across 400+ million structures also makes it a benchmarking study for evaluating current chemoinformatics tools.
The Tautomerism Problem in Chemical Databases
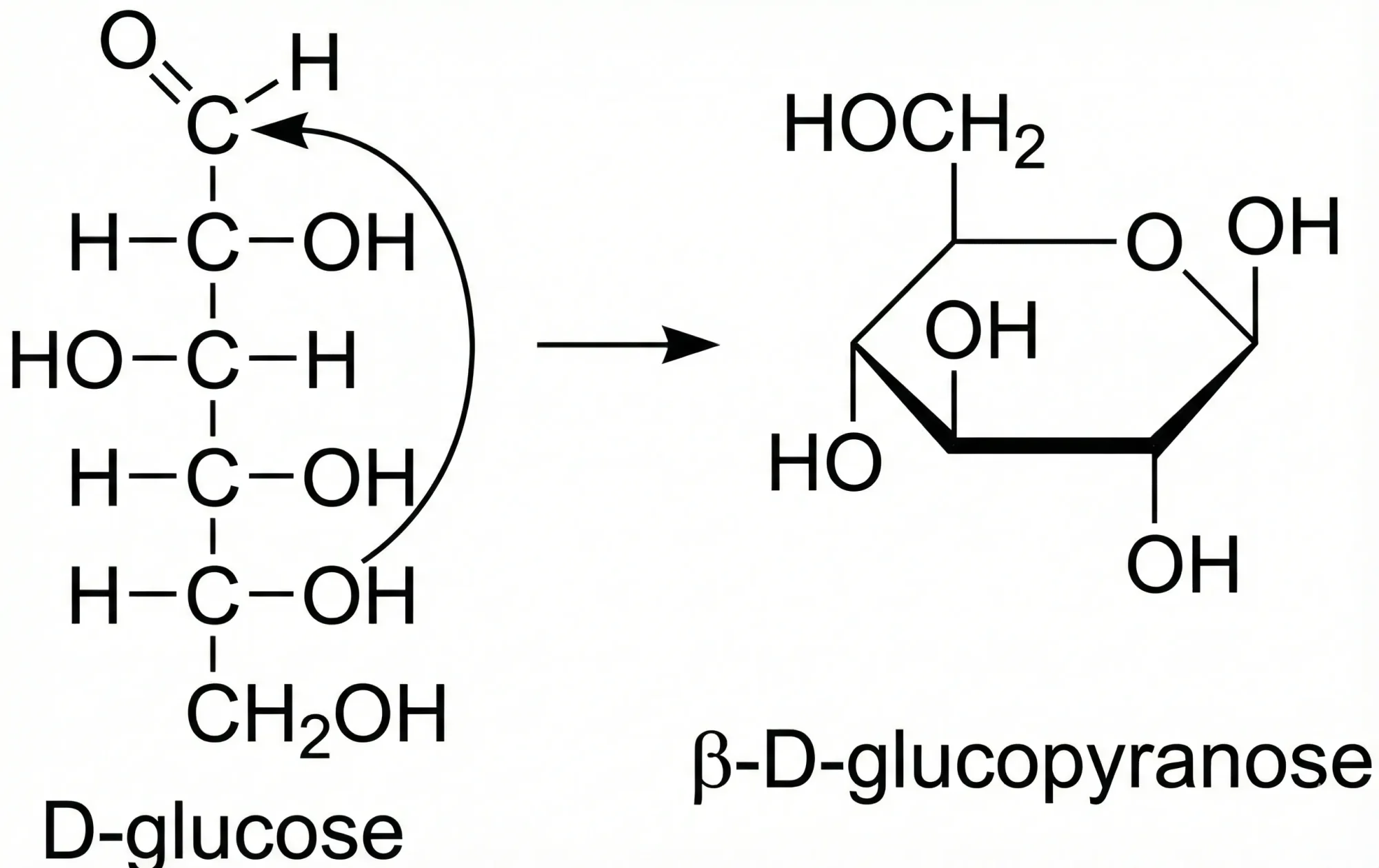
Chemical databases face a fundamental problem: the same molecule can appear multiple times under different identifiers simply because it exists in different tautomeric forms. For example, glucose’s ring-closed and open-chain forms are the same molecule; however, current chemical identifiers (including InChI) often treat them as distinct compounds.

This creates three critical problems:
- Database redundancy: Millions of duplicate entries for the same chemical entities
- Search failures: Researchers miss relevant compounds during structure searches
- ML training issues: Machine learning models learn to treat tautomers as different molecules
The motivation for this work is to provide a comprehensive, experimentally-grounded rule set that enables InChI V2 to properly recognize tautomeric relationships, eliminating these problems at the identifier level.
86 Comprehensive Tautomeric Transformation Rules
The key contributions are:
Comprehensive Rule Set: Compilation of 86 tautomeric transformation rules (20 pre-existing CACTVS defaults plus 66 new rules derived from experimental literature), categorized into:
- 54 Prototropic rules (classic H-movement tautomerism)
- 21 Ring-Chain rules (cyclic/open-chain transformations)
- 11 Valence rules (structural rearrangements with valence changes)
Massive-Scale Validation: Testing these rules against nine major chemical databases totaling over 400 million structures to identify coverage gaps in current InChI implementations
Quantitative Assessment: Systematic measurement showing that current InChI (even with Nonstandard 15T + KET settings) only achieves ~50% success in recognizing tautomeric relationships, with some new rules showing <2% success rates
Practical Tools: Creation of the Tautomerizer web tool for public use, demonstrating practical application of the rule set
The novelty lies in the systematic compilation and validation of transformation rules at a scale that reveals critical gaps in current chemical identification systems.
Massive-Scale Validation Across 400M+ Structures
Database Analysis
The researchers analyzed 9 chemical databases totaling 400+ million structures:
- Public databases: PubChem (largest), ChEMBL, DrugBank, PDB Ligands, SureChEMBL, AMS, ChemNavigator
- Private databases: CSD (Cambridge Structural Database), CSDB (NCI internal)
Methodology
Software: CACTVS Chemoinformatics Toolkit (versions 3.4.6.33 and 3.4.8.6)
Tautomer Generation Protocol:
- Algorithm: Single-step generation (apply transforms to input structure only, avoiding recursion)
- Constraints: Max 10 tautomers per structure, 30-second CPU timeout per transform
- Format: All rules expressed as SMIRKS strings
- Stereochemistry: Stereocenters involved in tautomerism were flattened during transformation
Success Metrics (tested against InChI V.1.05):
- Complete InChI match: All tautomers share identical InChI
- Partial InChI match: At least two tautomers share an InChI
- Tested against two InChI configurations: Standard InChI and Nonstandard InChI (with 15T and KET options enabled)
Rule Coverage Analysis
For each of the 86 rules, the researchers:
- Applied the transformation to all molecules in each database
- Generated tautomers using the SMIRKS patterns
- Computed InChI identifiers for each tautomer
- Measured success rates (percentage of cases where InChI recognized the relationship)
Key Findings from Experiments
Rule Frequency: The most common rule PT_06_00 (1,3-heteroatom H-shift, covering keto-enol tautomerism) affects >70% of molecules across databases.
InChI Performance:
- Standard InChI: ~37% success rate
- Nonstandard InChI (15T + KET): ~50% success rate
- Many newly defined rules: <2% success rate
Scale Impact: Implementing the full 86-rule set would approximately triple the number of compounds recognized as having tautomeric relationships relative to Standard InChI.
Outcomes: InChI V2 Requirements and Coverage Gaps
Main Findings
Current Systems Are Inadequate: Even with the Nonstandard 15T + KET settings, InChI only achieves ~50% success in recognizing tautomeric relationships, with Standard InChI at ~37%
Massive Coverage Gap: The new rule set reveals millions of tautomeric relationships that current InChI completely misses, particularly for ring-chain and valence tautomerism
Implementation Requirement: InChI V2 will require a major redesign to handle the comprehensive rule set
Rule Validation: The 86-rule set provides a validated foundation for next-generation chemical identifiers, with the new rules further confirmed against an independent ChEMBL 24.1 tautomer extraction
Implications
For Chemical Databases:
- Reduced redundancy through proper tautomer recognition
- Improved data quality and consistency
- More comprehensive structure search results
For Machine Learning:
- More accurate training data (tautomers properly grouped)
- Better molecular property prediction models
- Reduced dataset bias from tautomeric duplicates
For Chemoinformatics Tools:
- Blueprint for InChI V2 development
- Standardized rule set for tautomer generation
- Public tool (Tautomerizer) for practical use
Limitations Acknowledged
- Single-step generation only (omits recursive enumeration of all possible tautomers)
- 30-second timeout may miss complex transformations
- Some tautomeric preferences are context-dependent (pH, solvent) and require more than static rules for capture
Additional Validation
The authors validated their rule set against 4,158 tautomeric systems independently extracted from ChEMBL 24.1 via a SMILES-based tautomer hash (provided by Noel O’Boyle and Roger Sayle). Their rules covered essentially all tautomeric systems in that set, with practically all cases handled by the standard CACTVS rules PT_02_00 through PT_21_00.
Companion Resource: Tautomer Database
A companion paper describes the creation of a publicly available Tautomer Database (Tauto DB) containing over 2,800 tautomeric tuples extracted from experimental literature, available at https://cactus.nci.nih.gov/download/tautomer/. Data from this database informed the generation of new rules in this work.
Future Directions
The paper lays groundwork for InChI V2 development, emphasizing that the comprehensive rule set necessitates algorithmic redesign.
Reproducibility Details
Data
Datasets Analyzed (400M+ total structures):
Public Databases (Enable partial reproduction):
- PubChem: Largest public chemical database
- ChEMBL: Bioactive molecules with drug-like properties
- DrugBank: FDA-approved and experimental drugs
- PDB Ligands: Small molecules from protein structures
- SureChEMBL: Chemical structures from patents
- AMS: Screening samples
- ChemNavigator: Commercial chemical database
Private/Proprietary Databases (Prevent 100% full-scale reproduction):
- CSD: Cambridge Structural Database (requires commercial/academic license)
- CSDB: NCI internal database (private)
Algorithms
Tautomer Generation:
- Method: Single-step SMIRKS-based transformations
- Constraints:
- Maximum 10 tautomers per input structure
- 30-second CPU timeout per transformation
- Stereochemistry flattening for affected centers
- Toolkit Dependency: The authors used the CACTVS Chemoinformatics Toolkit. Researchers attempting to reproduce this with fully open-source tools (like RDKit) may encounter differing behavior due to proprietary chemical perception logic and licensing differences.
Rule Categories:
- Prototropic (PT): 54 rules for hydrogen movement
- Most common:
PT_06_00(1,3-heteroatom H-shift, >70% coverage)
- Most common:
- Ring-Chain (RC): 21 rules for cyclic/open-chain transformations
- Examples:
RC_03_00(pentose sugars),RC_04_01(hexose sugars)
- Examples:
- Valence (VT): 11 rules for valence changes
- Notable:
VT_02_00(tetrazole/azide, ~2.8M hits)
- Notable:
InChI Comparison:
- Standard InChI (default settings)
- Nonstandard InChI with
15TandKEToptions (mobile H and keto-enol)
Evaluation
Success Metrics:
Let $\mathcal{T}(m)$ be the set of generated tautomers for molecule $m$.
- Complete Match: Occurs iff $\forall t_i, t_j \in \mathcal{T}(m), \text{InChI}(t_i) = \text{InChI}(t_j)$.
- Partial Match: At least 2 tautomers share the same InChI.
- Fail: All tautomers have different InChIs.
Benchmark Results:
- Standard InChI: ~37% success rate across all rules
- Nonstandard (15T + KET): ~50% success rate
- New rules: Many show <2% recognition by current InChI
Hardware
Software Environment:
- Toolkit: CACTVS Chemoinformatics Toolkit v3.4.6.33 and v3.4.8.6
- Hash Functions:
E_TAUTO_HASH(tautomer-invariant identifier)E_ISOTOPE_STEREO_HASH128(tautomer-sensitive identifier)
Note: The paper omits computational hardware specifications but acknowledges using the NIH HPC Biowulf cluster. Evaluating 400M+ structures necessitates high-throughput cluster computing, making it computationally expensive for an individual to replicate the full analysis from scratch.
Artifacts
| Artifact | Type | License | Notes |
|---|---|---|---|
| Tautomerizer Web Tool | Other | Unknown | Public web tool for applying tautomeric rules to user molecules |
| Tautomer Database | Dataset | Unknown | 2800+ experimental tautomeric tuples (companion resource) |
| SMIRKS and Scripts (SI) | Code | Unknown | CACTVS Tcl scripts and SMIRKS provided as Supporting Information |
Paper Information
Citation: Dhaked, D. K., Ihlenfeldt, W.-D., Patel, H., Delannée, V., & Nicklaus, M. C. (2020). Toward a Comprehensive Treatment of Tautomerism in Chemoinformatics Including in InChI V2. Journal of Chemical Information and Modeling, 60(3), 1253-1275. https://doi.org/10.1021/acs.jcim.9b01080
Publication: Journal of Chemical Information and Modeling, 2020
@article{dhaked2020toward,
title={Toward a Comprehensive Treatment of Tautomerism in Chemoinformatics Including in InChI V2},
author={Dhaked, Devendra K and Ihlenfeldt, Wolf-Dietrich and Patel, Hitesh and Delann{\'e}e, Victorien and Nicklaus, Marc C},
journal={Journal of Chemical Information and Modeling},
volume={60},
number={3},
pages={1253--1275},
year={2020},
publisher={ACS Publications},
doi={10.1021/acs.jcim.9b01080}
}
Additional Resources:
- Tautomerizer Tool - Public web tool for testing tautomeric transformations