A Fragment-Aware Extension of SELFIES
This is a Method paper that introduces Group SELFIES, a molecular string representation extending SELFIES by incorporating group tokens that represent functional groups or entire substructures. The primary contribution is a representation that maintains the 100% chemical validity guarantee of SELFIES while enabling fragment-level molecular encoding. Group SELFIES is shorter, more human-readable, and produces better distribution learning compared to both SMILES and standard SELFIES.
From Atoms to Fragments in Molecular Strings
Molecular string representations underpin nearly all string-based molecular generation, from chemical language models and VAEs to genetic algorithms. SMILES, the dominant representation, suffers from validity issues: generated strings frequently contain syntax errors or violate valency constraints. SELFIES solved this by guaranteeing that every string decodes to a valid molecule, but both SMILES and SELFIES operate at the atomic level. Human chemists, by contrast, think about molecules in terms of functional groups and substructures.
Fragment-based generative models exploit this inductive bias by constructing custom representations amenable to fragment-based molecular design. However, these approaches are typically graph-based, losing the desirable properties of string representations: easy manipulation and direct input into established language models. Historical string representations like Wiswesser Line Notation (WLN), Hayward Notation, and SYBYL Line Notation (SLN) did use non-atomic tokens, but none provided chemical robustness guarantees.
The gap is clear: no existing string representation combines the chemical robustness of SELFIES with the fragment-level abstraction that captures meaningful chemical motifs.
Group Tokens with Chemical Robustness Guarantees
The core innovation is the introduction of group tokens into the SELFIES framework. Each group token represents a predefined molecular fragment (such as a benzene ring, carboxyl group, or any user-specified substructure) and is treated as a single unit during encoding and decoding.
Group Definition
Each group is defined as a set of atoms and bonds with labeled attachment points that specify how the group participates in bonding. Each attachment point has a specified maximum valency, allowing the decoder to continue tracking available valency during string construction. Group tokens take the form [:S<group-name>], where S is the starting attachment index.
Encoding
To encode a molecule, the encoder first recognizes and replaces substructure matches from the group set. By default, the encoder processes larger groups first, but users can override this with priority values. The encoder then traverses the molecular graph similarly to standard SELFIES encoding, inserting tokens that track attachment indices for entering and exiting groups.
Decoding
When the decoder encounters a group token, it looks up the corresponding group in the group set dictionary, places all atoms of the group, and connects the main chain to the starting attachment point. Navigation between attachment points is handled by reading subsequent tokens as relative indices. If an attachment point is occupied, the next available one is used. If all attachment points are exhausted, the group is immediately popped from the stack.
Chemical Robustness
The key property preserved from SELFIES is that any arbitrary Group SELFIES string decodes to a molecule with valid valency. This is achieved by maintaining the same two SELFIES decoder features within the group framework:
- Token overloading: every token can be interpreted as a number when needed (for branch lengths, ring targets, or attachment indices).
- Valency tracking: if adding a bond would exceed available valency, the decoder adjusts the bond order or skips the bond.
The authors verified robustness by encoding and decoding 25 million molecules from the eMolecules database.
Chirality Handling
Group SELFIES handles chirality differently from SMILES and SELFIES. Rather than using @-notation for tetrahedral chirality, all chiral centers must be specified as groups. An “essential set” of 23 groups covers all relevant chiral centers in the eMolecules database. This approach also supports extended chirality (axial, helical, planar) by abstracting the entire chiral substructure into a group token.
Fragment Selection
The group set is a user-defined dictionary that maps group names to molecular fragments. Users can specify groups manually using SMILES-like syntax, extract them from fragment libraries, or use fragmentation algorithms such as matched molecular pair analysis. The authors tested several approaches, including a naive method that cleaves side chains from rings and methods based on cheminformatics fragmentation tools. A useful group set typically contains fragments that appear in many molecules and replace many atoms, with similar fragments merged to reduce redundancy.
Experiments on Compactness, Generation, and Distribution Learning
Compactness (Section 4.1)
Using 53 groups (30 extracted from ZINC-250k plus 23 from the essential set), Group SELFIES strings are shorter than their SMILES and SELFIES equivalents. Despite Group SELFIES having a larger alphabet, the compressed file size of the ZINC-250k dataset is smallest for Group SELFIES, indicating lower information-theoretic complexity.
Random Molecular Generation (Section 4.2)
To isolate the effect of the representation from the generative model, the authors use a primitive generative model: sample a random string length from the dataset, draw tokens uniformly from a bag of all tokens, and concatenate. From 100,000 ZINC-250k molecules:
- Randomly sampled Group SELFIES strings produce molecules whose SAScore and QED distributions more closely overlap with the original ZINC dataset than molecules from randomly sampled SELFIES strings.
- The Wasserstein distances to the ZINC distribution are consistently lower for Group SELFIES.
- On a nonfullerene acceptor (NFA) dataset, Group SELFIES preserves aromatic rings while SELFIES rarely does.
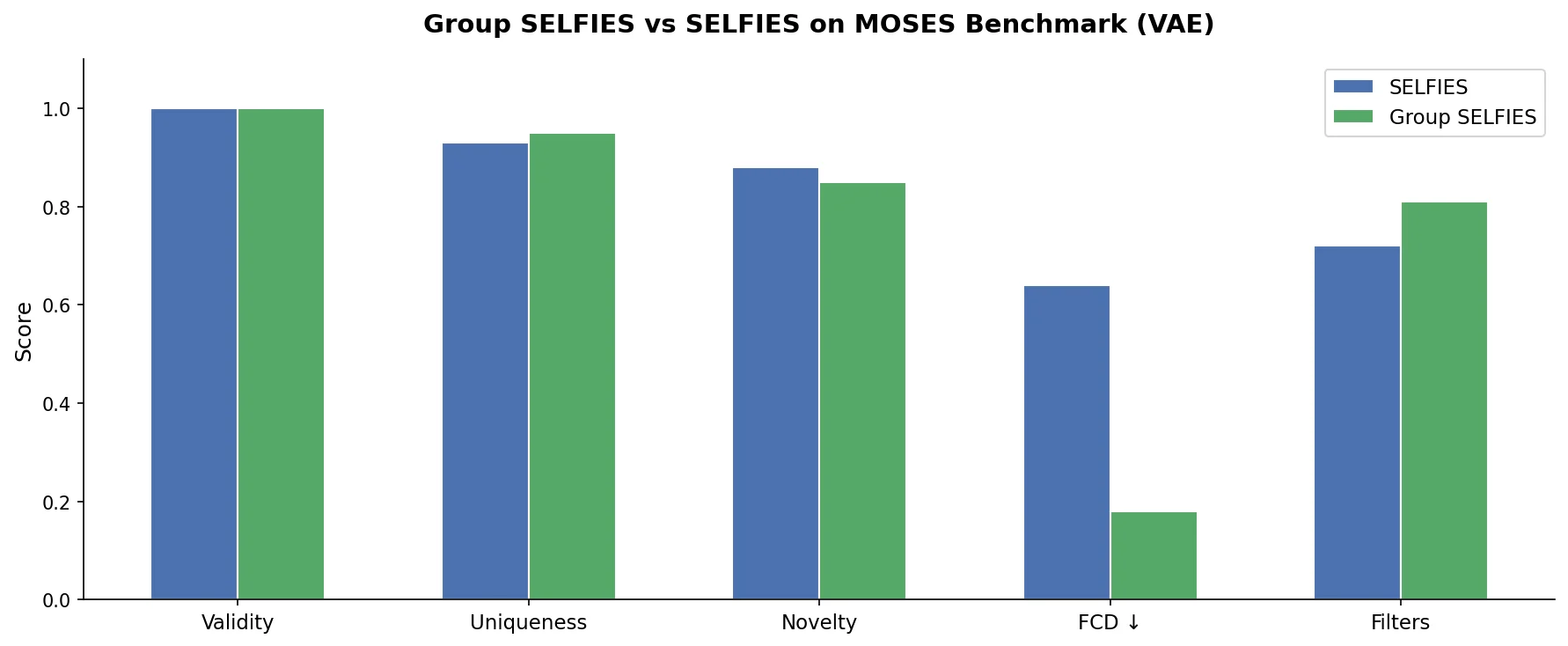
Distribution Learning with VAEs (Section 4.3)
Using the MOSES benchmarking framework, VAEs were trained for 125 epochs on both Group SELFIES and SELFIES representations. The Group SELFIES VAE used 300 groups extracted from the MOSES training set. Results from 100,000 generated molecules:
| Metric | Group-VAE-125 | SELFIES-VAE-125 | Train (Reference) |
|---|---|---|---|
| Valid | 1.0 (0) | 1.0 (0) | 1.0 |
| Unique@1k | 1.0 (0) | 0.9996 (5) | 1.0 |
| Unique@10k | 0.9985 (4) | 0.9986 (4) | 1.0 |
| FCD (Test) | 0.1787 (29) | 0.6351 (43) | 0.008 |
| FCD (TestSF) | 0.734 (109) | 1.3136 (128) | 0.4755 |
| SNN (Test) | 0.6051 (4) | 0.6014 (3) | 0.6419 |
| Frag (Test) | 0.9995 (0) | 0.9989 (0) | 1.0 |
| Scaf (Test) | 0.9649 (21) | 0.9588 (15) | 0.9907 |
| IntDiv | 0.8587 (1) | 0.8579 (1) | 0.8567 |
| Novelty | 0.9623 (7) | 0.96 (4) | 1.0 |
The most notable improvement is in Frechet ChemNet Distance (FCD), where Group SELFIES achieves 0.1787 versus 0.6351 for SELFIES on the test set. FCD measures the difference between penultimate-layer activations of ChemNet, encoding a mixture of biological and chemical properties relevant to drug-likeness. Most other metrics are comparable, with Group SELFIES matching or slightly outperforming SELFIES across the board.
Advantages, Limitations, and Future Directions
Key Findings
Group SELFIES provides three main advantages over standard SELFIES:
- Substructure control: Important scaffolds, chiral centers, and charged groups can be preserved during molecular optimization.
- Compactness: Group tokens represent multiple atoms, yielding shorter strings with lower information-theoretic complexity.
- Improved distribution learning: The FCD metric shows substantial improvement, indicating generated molecules better capture biological and chemical properties of the training set.
Both SELFIES and Group SELFIES achieve 100% validity, eliminating the validity issues associated with SMILES-based generation.
Limitations
The authors acknowledge several limitations:
- Computational speed: Encoding and decoding is slower than SELFIES due to RDKit overhead, particularly for the encoder which performs substructure matching for every group in the set.
- No group overlap: Groups cannot overlap in the current formulation, which limits expressiveness for polycyclic compounds.
- Group set design: Choosing an effective group set remains an open design choice that may require domain expertise or fragmentation algorithm tuning.
- Limited generative model evaluation: The paper focuses on random sampling and VAEs; evaluation with more sophisticated models (GANs, reinforcement learning, genetic algorithms) is left to future work.
Future Directions
The authors propose several extensions: flexible scaffold tokens that preserve topology while allowing atom-type variation, representations based on cellular complexes or hypergraphs to handle overlapping groups, and integration with genetic algorithms like JANUS for molecular optimization.
Reproducibility Details
Data
| Purpose | Dataset | Size | Notes |
|---|---|---|---|
| Compactness / Generation | ZINC-250k | 250,000 molecules | Random subset of 10,000 for fragment extraction; 100,000 for generation |
| Distribution Learning | MOSES benchmark | ~1.9M molecules | Standard train/test split from MOSES framework |
| Robustness Verification | eMolecules | 25M molecules | Full database encode-decode round trip |
| NFA Generation | NFA dataset | Not specified | Nonfullerene acceptors from Lopez et al. (2017) |
Algorithms
- Fragmentation: Naive ring-sidechain cleavage, matched molecular pair analysis, and diversity-based selection of 300 groups for VAE experiments.
- Essential set: 23 chiral groups covering all relevant chiral centers in eMolecules.
- Random generation: Bag-of-tokens sampling with length matched to dataset distribution.
Models
- VAE: Trained for 125 epochs on MOSES dataset using both SELFIES and Group SELFIES tokenizations.
- Architecture details follow the MOSES benchmark VAE configuration.
Evaluation
| Metric | Description |
|---|---|
| FCD | Frechet ChemNet Distance (penultimate layer activations) |
| SNN | Average Tanimoto similarity to nearest neighbor in reference set |
| Frag | Cosine similarity of BRICS fragment distributions |
| Scaf | Cosine similarity of Bemis-Murcko scaffold distributions |
| IntDiv | Internal diversity via Tanimoto similarity |
| Validity | Percentage passing RDKit parsing |
| Uniqueness | Percentage of non-duplicate generated molecules |
| Novelty | Fraction of generated molecules not in training set |
Hardware
- Robustness verification performed on the Niagara supercomputer (SciNet HPC Consortium).
- VAE training hardware not specified.
Artifacts
| Artifact | Type | License | Notes |
|---|---|---|---|
| group-selfies | Code | Apache-2.0 | Open-source Python implementation |
Paper Information
Citation: Cheng, A. H., Cai, A., Miret, S., Malkomes, G., Phielipp, M., & Aspuru-Guzik, A. (2023). Group SELFIES: A robust fragment-based molecular string representation. Digital Discovery, 2(3), 748-758. https://doi.org/10.1039/D3DD00012E
@article{cheng2023group,
title={Group SELFIES: A Robust Fragment-Based Molecular String Representation},
author={Cheng, Austin H. and Cai, Andy and Miret, Santiago and Malkomes, Gustavo and Phielipp, Mariano and Aspuru-Guzik, Al{\'a}n},
journal={Digital Discovery},
volume={2},
number={3},
pages={748--758},
year={2023},
publisher={Royal Society of Chemistry},
doi={10.1039/D3DD00012E}
}