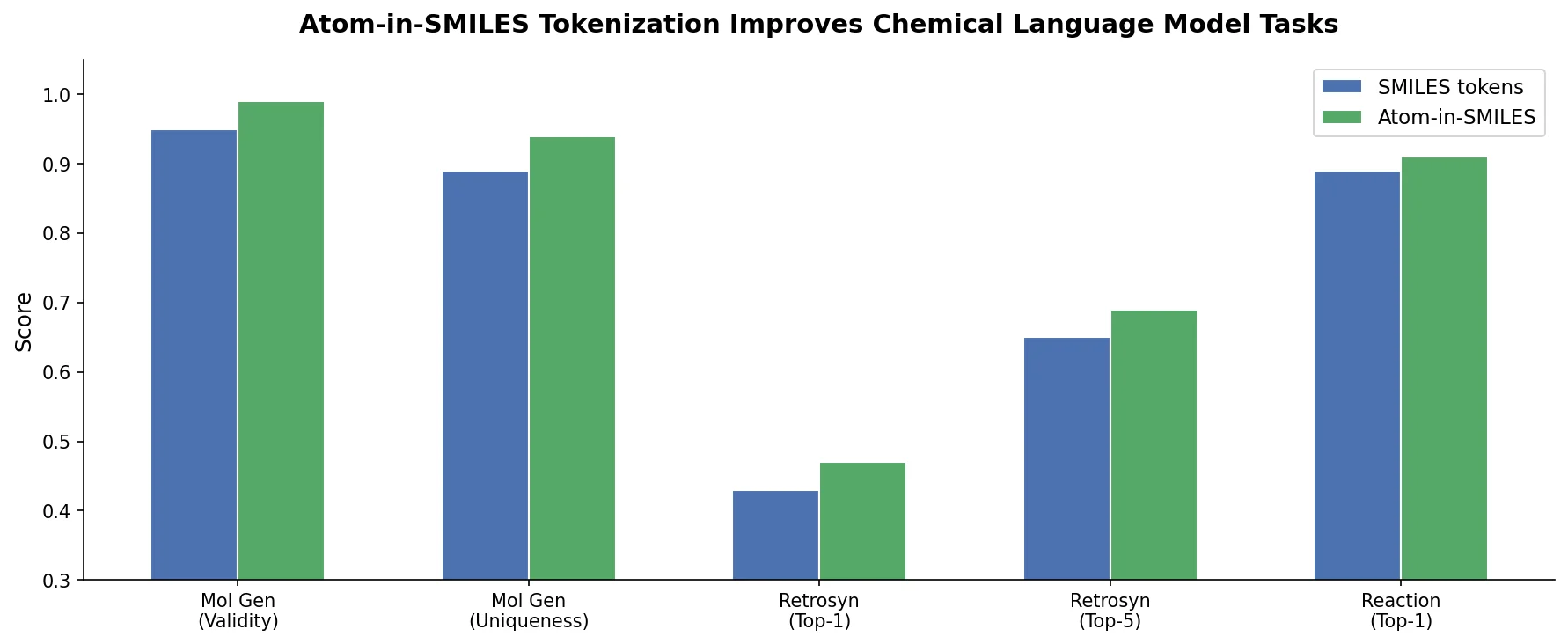
A New Tokenization Method for Chemical Language Models
This is a Method paper that introduces Atom-in-SMILES (AIS), a tokenization scheme for SMILES strings that replaces generic atomic tokens with environment-aware tokens encoding each atom’s local chemical neighborhood. The primary contribution is demonstrating that tokenization quality has a significant impact on chemical language model outcomes across multiple tasks: SMILES canonicalization, single-step retrosynthesis, and molecular property prediction.
Why Standard SMILES Tokenization Falls Short
Standard atom-wise SMILES tokenization treats all atoms of the same element identically. Every carbon is tokenized as “C” regardless of whether it is part of an aromatic ring, a carbonyl group, or a methyl chain. This creates a highly degenerate token space where chemically distinct atoms share the same representation.
The authors draw an analogy between natural language and chemical language. A typical SMILES sequence is about three times longer than a natural language sentence, yet the token vocabulary is roughly 1000 times smaller. This mismatch leads to extreme token repetition: the same tokens (C, c, N, O) appear many times within a single sequence. In natural language processing, token degeneration (where models repeatedly predict the same token) is a known failure mode of autoregressive decoders. The repetitive nature of SMILES tokens exacerbates this problem in chemical language models.
SMILES also lacks a one-to-one correspondence between tokens and chemical meaning. Two molecules that differ in only one atom substitution (e.g., swapping a carbon for a nitrogen in a ring) produce identical token sets under atom-wise tokenization, making it harder for models to distinguish structurally similar molecules.
Core Innovation: Encoding Atom Environments into Tokens
The key insight is to replace each atomic token with a richer token that encodes the atom’s local chemical environment, inspired by the atoms-in-molecules (AIM) concept from quantum chemistry. For a given SMILES string, the AIS mapping function $f$ operates on the token space:
$$ f(X) = \begin{cases} AE|_{X_{\text{central}}} & \text{if } X \text{ is an atom} \\ X & \text{otherwise} \end{cases} $$
where $AE|_{X_{\text{central}}}$ denotes the atomic environment centered on atom $X$. Non-atomic tokens (brackets, bond symbols, ring closures) pass through unchanged.
Each AIS token is formatted as [Sym;Ring;Neighbors] where:
- Sym is the atomic symbol with chirality, aromaticity (lowercase for aromatic), hydrogen count, and formal charge
- Ring indicates whether the atom is in a ring (
R) or not (!R) - Neighbors lists the neighboring atoms interacting with the central atom
This mapping is bijective: SMILES strings can be fully recovered from AIS strings via an inverse projection. The algorithm iterates over atoms in a molecule, computes their local environments using RDKit, and produces environment-aware token variants.
As a concrete example, in glycine the two carbons and two oxygens are indistinguishable under atom-wise tokenization. Under AIS, each receives a unique token reflecting its bonding environment (e.g., the carboxyl carbon is distinguished from the alpha carbon).
The AIS tokenization also exhibits a fingerprint-like property. Because each token encodes local structural information, the set of AIS tokens for a molecule functions similarly to circular fingerprints like ECFP2. The authors show that pairwise Tanimoto similarities computed from AIS token sets have resolution comparable to ECFP2 and HashAP fingerprints, and better resolution than MACCS, Avalon, and RDKit fingerprints.
Token repetition can be quantified as:
$$ \text{rep-}l = \sum_{t=1}^{|s|} \mathbb{1}[s_t \in s_{t-w-1:t-1}] $$
where $s$ is the predicted sequence, $|s|$ is the token count, and $w$ is the window size. AIS tokens exhibit consistently lower normalized repetition rates compared to SMILES, SELFIES, and DeepSMILES across diverse molecular datasets (drugs, natural products, steroids, lipids, metal complexes, octane isomers).
Experimental Evaluation Across Three Chemical Tasks
Input-Output Equivalent Mapping (SMILES Canonicalization)
The first task tests whether a model can translate non-canonical SMILES enumerations into canonical form. The authors constructed deliberately challenging datasets from GDB-13 subsets with cumulative structural constraints (no cyclic heteroatom-heteroatom bonds, stable functional groups only, fragment-like, scaffold-like, etc.), generating training sets of 1M molecules augmented with 150K molecules from the most restrictive subset at 10x, 30x, and 50x augmentation levels.
| GDB-13 Subset | Atom-wise (x10) | Atom-wise (x50) | AIS (x10) | AIS (x50) |
|---|---|---|---|---|
| ab | 34.2% | 33.2% | 37.3% | 34.1% |
| abc | 31.0% | 29.6% | 33.7% | 30.4% |
| abcde | 48.7% | 45.5% | 53.6% | 47.0% |
| abcdef | 41.8% | 39.1% | 52.5% | 46.9% |
| abcdefg | 50.9% | 50.0% | 59.9% | 56.8% |
AIS outperformed atom-wise tokenization on all subsets and augmentation levels. The performance gap grew larger for more restrictive (more similar) subsets, reaching up to 10.7% on the abcdef subset. This demonstrates that AIS is particularly effective when molecules are structurally similar and harder to distinguish.
Single-Step Retrosynthesis
The second task uses the USPTO-50K benchmark for single-step retrosynthetic prediction via a template-free transformer encoder-decoder model. The model was trained for 200,000 steps with Adam optimizer, negative log-likelihood loss, and cyclic learning rate scheduling.
| Tokenization | rep-|P - rep-|GT >= 2 | String Exact (%) | Tc Exact (%) |
|---|---|---|---|
| Atom-wise baseline | – | 42.00 | – |
| Atom-wise (reproduced) | 801 | 42.05 | 44.72 |
| SmilesPE | 821 | 19.82 | 22.74 |
| SELFIES | 886 | 28.82 | 30.76 |
| DeepSMILES | 902 | 38.63 | 41.20 |
| Atom-in-SMILES | 727 | 46.32 | 47.62 |
AIS achieved 46.32% string exact accuracy (4.3% above the atom-wise baseline) and 47.62% Tanimoto exact accuracy (2.9% above baseline). AIS also had the fewest degenerate token repetitions (727 vs. 801 for atom-wise), representing approximately a 10% reduction. DeepSMILES had the highest repetition count (902) despite reasonable overall accuracy. SELFIES and SmilesPE both performed substantially worse than the atom-wise baseline on this task.
The authors identified six common token repetition patterns in retrosynthetic predictions: long head repetitions, long tail repetitions, repetitive rings, repetitive chains, and halogen repetitions on both aliphatic and aromatic carbons.
Molecular Property Prediction
The third task evaluates tokenization schemes on MoleculeNet benchmarks using Random Forest models with 5-fold cross-validation. AIS tokens were converted to fingerprint-like feature vectors.
| Dataset | SMILES | DeepSMILES | SELFIES | SmilesPE | AIS |
|---|---|---|---|---|---|
| Regression (RMSE, lower is better) | |||||
| ESOL | 0.628 | 0.631 | 0.675 | 0.689 | 0.553 |
| FreeSolv | 0.545 | 0.544 | 0.564 | 0.761 | 0.441 |
| Lipophilicity | 0.924 | 0.895 | 0.938 | 0.800 | 0.683 |
| Classification (ROC-AUC, higher is better) | |||||
| BBBP | 0.758 | 0.777 | 0.799 | 0.847 | 0.885 |
| BACE | 0.740 | 0.774 | 0.746 | 0.837 | 0.835 |
| HIV | 0.649 | 0.648 | 0.653 | 0.739 | 0.729 |
AIS achieved the best performance on all three regression datasets and two of three classification datasets. On ESOL, the RMSE improvement over standard SMILES was 12%. On lipophilicity, the improvement was 26%.
Key Findings: Better Tokens Yield Better Chemical Models
The main findings of this work are:
Tokenization significantly impacts chemical language model quality. The choice of tokenization scheme can change prediction accuracy by over 10 percentage points on equivalent mapping tasks.
AIS reduces token degeneration by approximately 10% compared to atom-wise SMILES tokenization, with consistently lower normalized repetition rates across diverse molecular datasets.
AIS outperforms all compared tokenization schemes (atom-wise SMILES, SmilesPE, SELFIES, DeepSMILES) on canonicalization, retrosynthesis, and property prediction.
The fingerprint-like nature of AIS tokens enables direct use as molecular features for property prediction and provides resolution comparable to established circular fingerprints.
The mapping is invertible, so AIS strings can always be converted back to valid SMILES. This is a practical advantage over approaches that may lose structural information.
Limitations: AIS cannot distinguish environmentally identical substructures or atoms related by a molecular symmetry plane, since it only considers nearest-neighbor environments. Performance on long-chain molecules (e.g., lipids) is similar across all tokenization schemes, suggesting that local environment encoding is less informative for repetitive linear structures.
Future directions: The authors suggest AIS has potential for broader adoption in molecular generative models, chemical translation, and property prediction tasks across the cheminformatics community.
Reproducibility Details
Data
| Purpose | Dataset | Size | Notes |
|---|---|---|---|
| Canonicalization training | GDB-13 subsets | 1M + 150K augmented | Cumulative structural constraints a-h |
| Canonicalization testing | GDB-13 disjoint test sets | 20K per subset | Various restriction levels |
| Retrosynthesis | USPTO-50K | ~50K reactions | Sequences > 150 tokens removed |
| Property prediction | MoleculeNet (ESOL, FreeSolv, Lipophilicity, BBBP, BACE, HIV) | Varies | Standard benchmark splits |
Algorithms
- Transformer encoder-decoder architecture for canonicalization and retrosynthesis tasks
- 200,000 training steps with Adam optimizer, negative log-likelihood loss, cyclic learning rate scheduler
- Random Forest with 5-fold cross-validation for property prediction
- AIS tokenization implemented via RDKit for atom environment extraction
Evaluation
| Metric | Task | Notes |
|---|---|---|
| String exact match (%) | Canonicalization, Retrosynthesis | Exact SMILES match |
| Tanimoto exactness (Tc) | Retrosynthesis | Morgan FP radius 3, 2048 bits |
| RMSE | Regression property prediction | ESOL, FreeSolv, Lipophilicity |
| ROC-AUC | Classification property prediction | BBBP, BACE, HIV |
| rep-l | Token degeneration | Single-token repetition count |
Hardware
Not explicitly specified in the paper.
Artifacts
| Artifact | Type | License | Notes |
|---|---|---|---|
| atom-in-SMILES | Code | CC-BY-NC-SA-4.0 | AIS tokenization implementation |
Paper Information
Citation: Ucak, U. V., Ashyrmamatov, I., & Lee, J. (2023). Improving the quality of chemical language model outcomes with atom-in-SMILES tokenization. Journal of Cheminformatics, 15, 55. https://doi.org/10.1186/s13321-023-00725-9
@article{ucak2023improving,
title={Improving the quality of chemical language model outcomes with atom-in-SMILES tokenization},
author={Ucak, Umit V. and Ashyrmamatov, Islambek and Lee, Juyong},
journal={Journal of Cheminformatics},
volume={15},
number={1},
pages={55},
year={2023},
publisher={Springer},
doi={10.1186/s13321-023-00725-9}
}