A Unified Framework for All-Atom Molecular Foundation Models
PharMolixFM is a Method paper that introduces a unified framework for constructing all-atom foundation models for molecular modeling and generation. The primary contribution is the systematic implementation of three multi-modal generative model variants (diffusion, flow matching, and Bayesian flow networks) within a single architecture, along with a task-unifying denoising formulation that enables training on multiple structural biology tasks simultaneously. The framework achieves competitive performance on protein-small-molecule docking and structure-based drug design while providing the first empirical analysis of inference scaling laws for molecular generative models.
Challenges in Multi-Modal Atomic Modeling
Existing all-atom foundation models such as AlphaFold3, RoseTTAFold All-Atom, and ESM-AA face two core challenges that limit their generalization across molecular modeling and generation tasks.
First, atomic data is inherently multi-modal: each atom comprises both a discrete atom type and continuous 3D coordinates. This poses challenges for structure models that need to jointly capture and predict both modalities. Unlike text or image data that exhibit a single modality, molecular structures require generative models that can handle discrete categorical variables (atom types, bond types) and continuous variables (coordinates) simultaneously.
Second, there has been no comprehensive analysis of how different training objectives and sampling strategies impact the performance of all-atom foundation models. Prior work has focused on individual model architectures without systematically comparing generative frameworks or studying how inference-time compute scaling affects prediction quality.
PharMolixFM addresses both challenges by providing a unified framework that implements three state-of-the-art multi-modal generative models and formulates all downstream tasks as a generalized denoising process with task-specific priors.
Multi-Modal Denoising with Task-Specific Priors
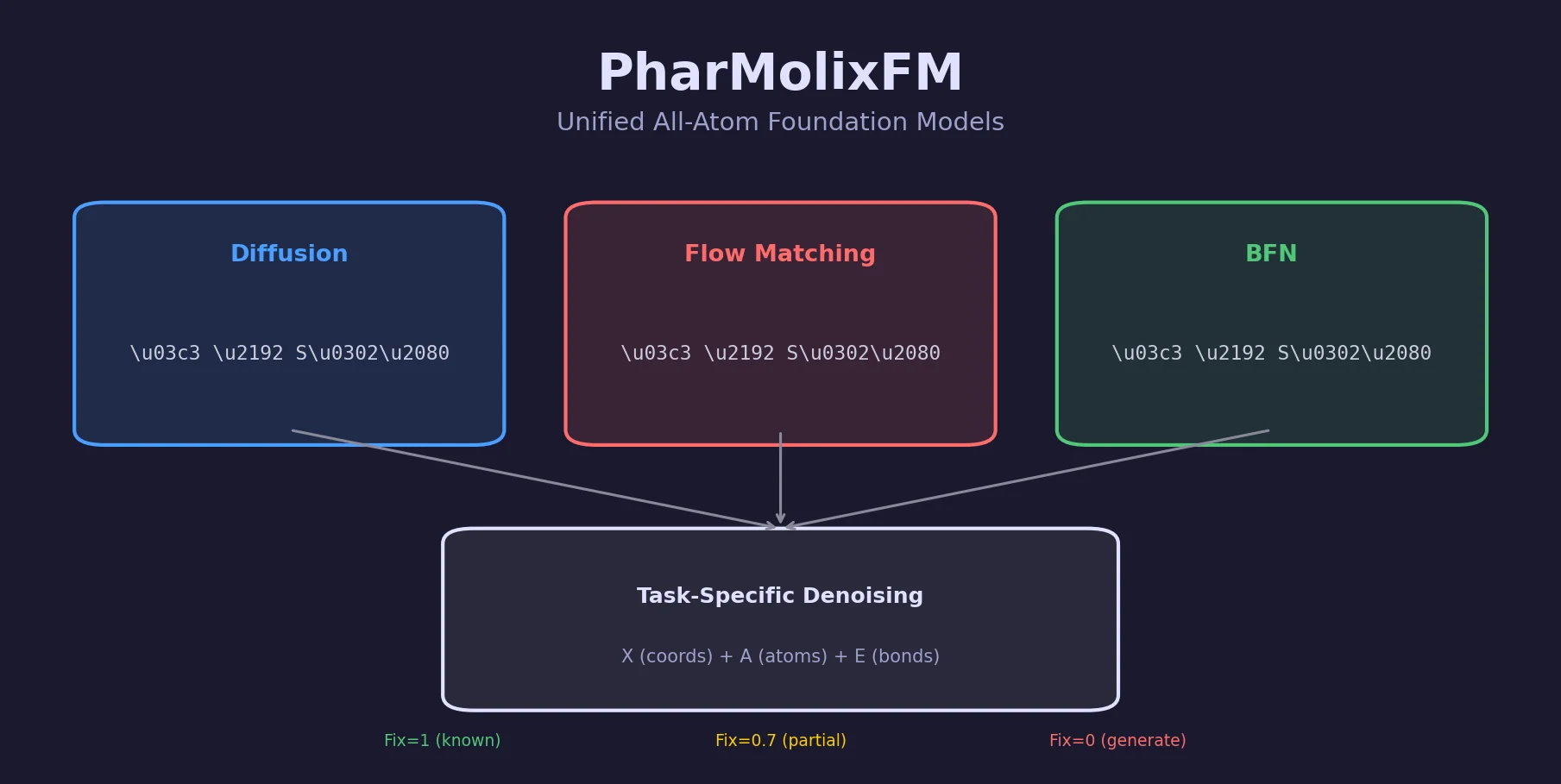
The core innovation of PharMolixFM is the formulation of molecular tasks as a generalized denoising process where task-specific priors control which parts of the molecular system are noised during training. The framework decomposes a biomolecular system into $N$ atoms represented as a triplet $\bar{\mathbf{S}}_0 = \langle \mathbf{X}_0, \mathbf{A}_0, \mathbf{E}_0 \rangle$, where $\mathbf{X}_0 \in \mathbb{R}^{N \times 3}$ are atom coordinates, $\mathbf{A}_0 \in \mathbb{Z}^{N \times D_1}$ are one-hot atom types, and $\mathbf{E}_0 \in \mathbb{Z}^{N \times N \times D_2}$ are one-hot bond types.
The generative model estimates the density $p_\theta(\langle \mathbf{X}_0, \mathbf{A}_0, \mathbf{E}_0 \rangle)$ subject to SE(3) invariance:
$$ p_\theta(\langle \mathbf{R}\mathbf{X}_0 + \mathbf{t}, \mathbf{A}_0, \mathbf{E}_0 \rangle) = p_\theta(\langle \mathbf{X}_0, \mathbf{A}_0, \mathbf{E}_0 \rangle) $$
The variational lower bound is optimized over latent variables $S_1, \ldots, S_T$ obtained by adding independent noise to different modalities and atoms:
$$ q(S_{1:T} \mid S_0) = \prod_{i=1}^{T} \prod_{j=1}^{N} q(\mathbf{X}_{i,j} \mid \mathbf{X}_{0,j}, \sigma_{i,j}^{(\mathbf{X})}) , q(\mathbf{A}_{i,j} \mid \mathbf{A}_{0,j}, \sigma_{i,j}^{(\mathbf{A})}) , q(\mathbf{E}_{i,j} \mid \mathbf{E}_{0,j}, \sigma_{i,j}^{(\mathbf{E})}) $$
A key design choice is the noise schedule $\sigma_{i,j}^{(\mathcal{M})} = \frac{i}{T} \cdot \text{fix}_j^{(\mathcal{M})}$, where $\text{fix}_j^{(\mathcal{M})}$ is a scaling factor between 0 and 1 that controls which atoms and modalities receive noise. This “Fix” mechanism enables multiple training tasks:
- Docking ($\text{Fix} = 1$ for protein and molecular graph, $\text{Fix} = 0$ for molecule coordinates): predicts binding pose given known atom/bond types.
- Structure-based drug design ($\text{Fix} = 1$ for protein, $\text{Fix} = 0$ for all molecule properties): generates novel molecules for a given pocket.
- Robustness augmentation ($\text{Fix} = 0.7$ for 15% randomly selected atoms, $\text{Fix} = 0$ for rest): simulates partial structure determination.
Three Generative Model Variants
Multi-modal diffusion (PharMolixFM-Diff) uses a Markovian forward process. Continuous coordinates follow Gaussian diffusion while discrete variables use a D3PM categorical transition:
$$ q(\mathbf{X}_{i,j} \mid \mathbf{X}_{0,j}) = \mathcal{N}(\sqrt{\alpha_{i,j}} , \mathbf{X}_{0,j}, (1 - \alpha_{i,j}) \mathbf{I}), \quad \alpha_{i,j} = \prod_{k=1}^{i}(1 - \sigma_{i,j}^{(\mathbf{X})}) $$
$$ q(\mathbf{A}_{i,j} \mid \mathbf{A}_{0,j}) = \text{Cat}(\mathbf{A}_{0,j} \bar{Q}_{i,j}^{(\mathbf{A})}), \quad Q_{i,j}^{(\mathbf{A})} = (1 - \sigma_{i,j}^{(\mathbf{A})}) \mathbf{I} + \frac{\sigma_{i,j}^{(\mathbf{A})}}{D_1} \mathbb{1}\mathbb{1}^T $$
The training loss combines coordinate MSE with cross-entropy for discrete variables:
$$ \mathcal{L} = \mathbb{E}_{S_0, i, S_i} \left[ \lambda_i^{(\mathbf{X})} | \tilde{\mathbf{X}}_0 - \mathbf{X}_0 |_2^2 + \lambda_i^{(\mathbf{A})} \mathcal{L}_{CE}(\tilde{\mathbf{A}}_0, \mathbf{A}_0) + \lambda_i^{(\mathbf{E})} \mathcal{L}_{CE}(\tilde{\mathbf{E}}_0, \mathbf{E}_0) \right] $$
Multi-modal flow matching (PharMolixFM-Flow) constructs a direct mapping between data and prior distributions using conditional vector fields. For coordinates, the conditional flow uses a Gaussian path $q(\mathbf{X}_{i,j} \mid \mathbf{X}_{0,j}) = \mathcal{N}((1 - \sigma_{i,j}^{(\mathbf{X})}) \mathbf{X}_{0,j}, (\sigma_{i,j}^{(\mathbf{X})})^2 \mathbf{I})$, while discrete variables use the same D3PM Markov chain. Sampling proceeds by solving an ODE via Euler integration.
Bayesian flow networks (PharMolixFM-BFN) perform generative modeling in the parameter space of the data distribution rather than the data space. The Bayesian flow distribution for coordinates is:
$$ p_F(\tilde{\mathbf{X}}_{i,j}^{(\theta)} \mid \mathbf{X}_{0,j}) = \mathcal{N}(\gamma_{i,j} \mathbf{X}_{0,j}, \gamma_{i,j}(1 - \gamma_{i,j}) \mathbf{I}), \quad \gamma_{i,j} = 1 - \alpha^{2(1 - \sigma_{i,j}^{(\mathbf{X})})} $$
Network Architecture
The architecture follows PocketXMol with a dual-branch SE(3)-equivariant graph neural network. A protein branch (4-layer GNN with kNN graph) processes pocket atoms, then representations are passed to a molecule branch (6-layer GNN) that captures protein-molecule interactions. Independent prediction heads reconstruct atom coordinates, atom types, and bond types, with additional confidence heads for self-ranking during inference.
Docking and Drug Design Experiments
Protein-Small-Molecule Docking
PharMolixFM is evaluated on the PoseBusters benchmark (428 protein-small-molecule complexes) using the holo docking setting with a known protein structure and 10 Angstrom binding pocket. The metric is the ratio of predictions with RMSD < 2 Angstrom.
| Method | Self-Ranking (%) | Oracle-Ranking (%) |
|---|---|---|
| DiffDock | 38.0 | - |
| RFAA | 42.0 | - |
| Vina | 52.3 | - |
| UniMol-Docking V2 | 77.6 | - |
| SurfDock | 78.0 | - |
| AlphaFold3 | 90.4 | - |
| PocketXMol (50 repeats) | 82.2 | 95.3 |
| PharMolixFM-Diff (50 repeats) | 83.4 | 96.0 |
| PharMolixFM-Flow (50 repeats) | 73.4 | 93.7 |
| PharMolixFM-BFN (50 repeats) | 78.5 | 93.5 |
| PharMolixFM-Diff (500 repeats) | 83.9 | 98.1 |
PharMolixFM-Diff achieves the second-best self-ranking result (83.4%), outperforming PocketXMol by 1.7% absolute but trailing AlphaFold3 (90.4%). The key advantage is inference speed: approximately 4.6 seconds per complex on a single A800 GPU compared to approximately 249.0 seconds for AlphaFold3 (a 54x speedup). Under oracle-ranking with 500 repeats, PharMolixFM-Diff reaches 98.1%, suggesting that better ranking strategies could further improve practical performance.
Structure-Based Drug Design
Evaluation uses the CrossDocked test set (100 protein pockets, 100 molecules generated per pocket), measuring Vina binding affinity scores and drug-likeness properties (QED and SA).
| Method | Vina Score (Avg/Med) | QED | SA |
|---|---|---|---|
| Pocket2Mol | -5.14 / -4.70 | 0.57 | 0.76 |
| TargetDiff | -5.47 / -6.30 | 0.48 | 0.58 |
| DecompDiff | -5.67 / -6.04 | 0.45 | 0.61 |
| MolCRAFT | -6.61 / -8.14 | 0.46 | 0.62 |
| PharMolixFM-Diff | -6.18 / -6.44 | 0.50 | 0.73 |
| PharMolixFM-Flow | -6.34 / -6.47 | 0.49 | 0.74 |
| PharMolixFM-BFN | -6.38 / -6.45 | 0.48 | 0.64 |
PharMolixFM achieves a better balance between binding affinity and drug-like properties compared to baselines. While MolCRAFT achieves the best Vina scores, PharMolixFM-Diff and Flow variants show notably higher QED (0.49-0.50 vs. 0.45-0.48) and SA (0.73-0.74 vs. 0.58-0.62), which are important for downstream validation and in-vivo application.
Inference Scaling Law
The paper explores whether inference-time scaling holds for molecular generative models, fitting the relationship:
$$ \text{Acc} = a \log(bR + c) + d $$
where $R$ is the number of sampling repeats. All three PharMolixFM variants exhibit logarithmic improvement in docking accuracy with increased sampling repeats, analogous to inference scaling laws observed in NLP. Performance plateaus eventually due to distributional differences between training and test sets.
Competitive Docking with Faster Inference, but Limited Task Scope
PharMolixFM demonstrates that multi-modal generative models can achieve competitive all-atom molecular modeling with substantial inference speed advantages over AlphaFold3. The key findings are:
- Diffusion outperforms flow matching and BFN for docking under standard sampling budgets. The stochastic nature of diffusion sampling appears beneficial compared to the deterministic ODE integration of flow matching.
- Oracle-ranking reveals untapped potential: the gap between self-ranking (83.4%) and oracle-ranking (98.1%) at 500 repeats indicates that confidence-based ranking is a bottleneck. Better ranking methods could close the gap with AlphaFold3.
- The three variants show similar performance for drug design, suggesting that model architecture and training data may matter more than the generative framework for generation tasks.
- Inference scaling laws hold for molecular generative models, paralleling findings in NLP.
Limitations include that the framework is only evaluated on two tasks (docking and SBDD), and the paper does not address protein structure prediction, protein-protein interactions, or nucleic acid modeling, which are part of AlphaFold3’s scope. The BFN variant underperforms the diffusion model, which the authors attribute to smaller noise scales at early sampling steps making training less challenging. The paper also does not compare against concurrent work on inference-time scaling for molecular models.
Reproducibility Details
Data
| Purpose | Dataset | Size | Notes |
|---|---|---|---|
| Training | PDBBind, Binding MOAD, CrossDocked2020, PepBDB | Not specified | Filtered by PocketXMol criteria |
| Docking eval | PoseBusters benchmark | 428 complexes | Holo docking with known protein |
| SBDD eval | CrossDocked test set | 100 pockets | 100 molecules per pocket |
Algorithms
- Three generative variants: multi-modal diffusion (D3PM), flow matching, Bayesian flow networks
- Task-specific noise via Fix mechanism (0, 0.7, or 1.0)
- Training tasks selected with equal probability per sample
- AdamW optimizer: weight decay 0.001, $\beta_1 = 0.99$, $\beta_2 = 0.999$
- Linear warmup to learning rate 0.001 over 1000 steps
- 180K training steps with batch size 40
Models
- Dual-branch SE(3)-equivariant GNN (protein: 4-layer, molecule: 6-layer)
- kNN graph construction for protein and protein-molecule interactions
- Independent prediction heads for coordinates, atom types, bond types
- Confidence heads for self-ranking during inference
Evaluation
| Metric | PharMolixFM-Diff | AlphaFold3 | Notes |
|---|---|---|---|
| RMSD < 2A self-ranking | 83.4% (50 rep) | 90.4% | PoseBusters docking |
| RMSD < 2A oracle-ranking | 98.1% (500 rep) | - | PoseBusters docking |
| Inference time (per complex) | ~4.6s | ~249.0s | Single A800 GPU |
| Vina score (avg) | -6.18 | - | CrossDocked SBDD |
Hardware
- Training: 4x 80GB A800 GPUs
- Inference benchmarked on single A800 GPU
Artifacts
| Artifact | Type | License | Notes |
|---|---|---|---|
| OpenBioMed (GitHub) | Code | MIT | Official implementation |
Paper Information
Citation: Luo, Y., Wang, J., Fan, S., & Nie, Z. (2025). PharMolixFM: All-Atom Foundation Models for Molecular Modeling and Generation. arXiv preprint arXiv:2503.21788.
@article{luo2025pharmolixfm,
title={PharMolixFM: All-Atom Foundation Models for Molecular Modeling and Generation},
author={Luo, Yizhen and Wang, Jiashuo and Fan, Siqi and Nie, Zaiqing},
journal={arXiv preprint arXiv:2503.21788},
year={2025}
}