Paper Contribution Type
This is a method paper with a supporting theoretical component. It introduces a new pre-training framework, DenoiseVAE, that challenges the standard practice of using fixed, hand-crafted noise distributions in denoising-based molecular representation learning.
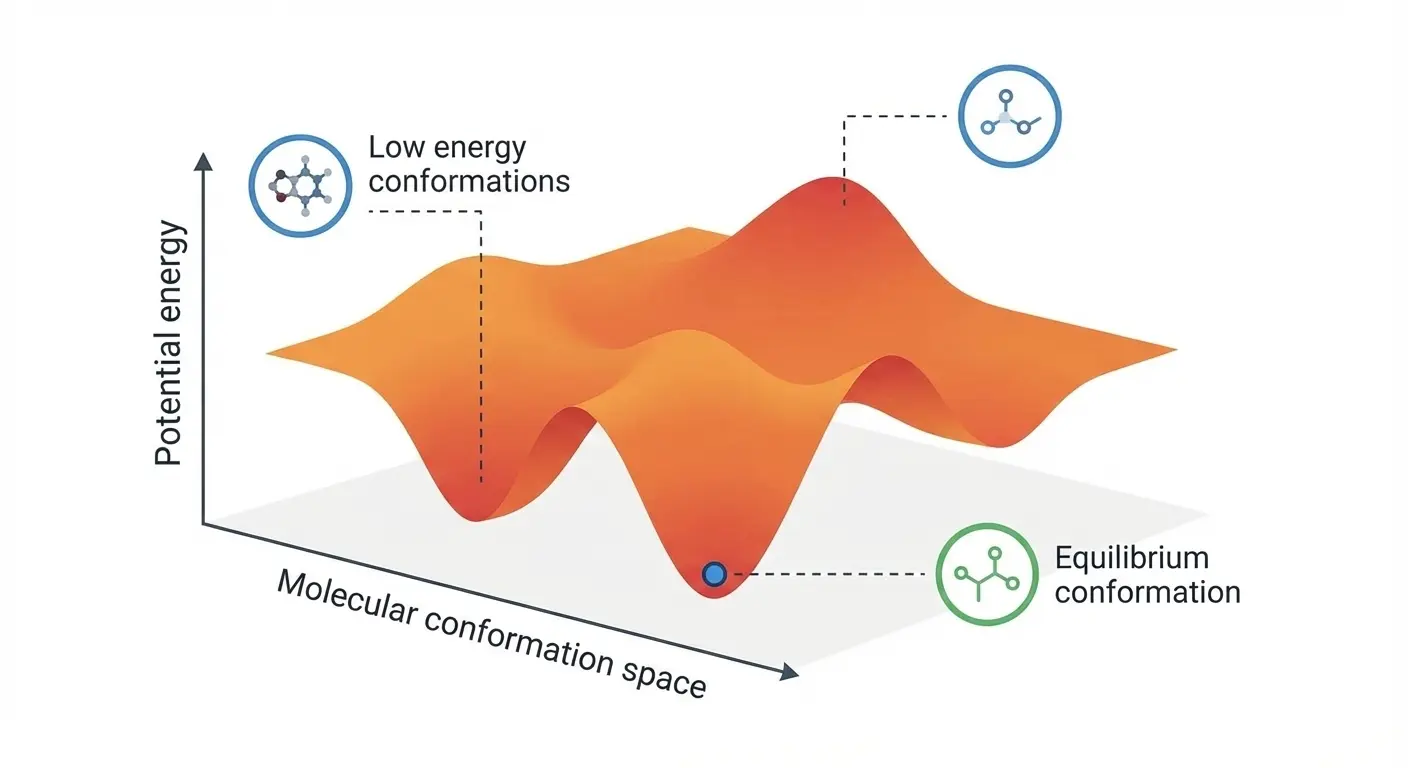
Motivation: The Inter- and Intra-molecular Variations Problem
The motivation is to create a more physically principled denoising pre-training task for 3D molecules. The core idea of denoising is to learn molecular force fields by corrupting an equilibrium conformation with noise and then learning to recover it. However, existing methods use a single, hand-crafted noise strategy (e.g., Gaussian noise of a fixed scale) for all atoms across all molecules. This is physically unrealistic for two main reasons:
- Inter-molecular differences: Different molecules have unique Potential Energy Surfaces (PES), meaning the space of low-energy (i.e., physically plausible) conformations is highly molecule-specific.
- Intra-molecular differences (Anisotropy): Within a single molecule, different atoms have different degrees of freedom. For instance, an atom in a rigid functional group can move much less than one connected by a single, rotatable bond.
The authors argue that this “one-size-fits-all” noise approach leads to inaccurate force field learning because it samples many physically improbable conformations.
Novelty: A Learnable, Atom-Specific Noise Generator
The core novelty is a framework that learns to generate noise tailored to each specific molecule and atom. This is achieved through three key innovations:
- Learnable Noise Generator: The authors introduce a Noise Generator module (a 4-layer Equivariant Graph Neural Network) that takes a molecule’s equilibrium conformation $X$ as input and outputs a unique, atom-specific Gaussian noise distribution (i.e., a different variance $\sigma_i^2$ for each atom $i$). This directly addresses the issues of PES specificity and force field anisotropy.
- Variational Autoencoder (VAE) Framework: The Noise Generator (encoder) and a Denoising Module (a 7-layer EGNN decoder) are trained jointly within a VAE paradigm. The noisy conformation is sampled using the reparameterization trick: $$ \begin{aligned} \tilde{x}_i &= x_i + \epsilon \sigma_i \end{aligned} $$
- Principled Optimization Objective: The training loss balances two competing goals:
$$
\begin{aligned}
\mathcal{L}_{DenoiseVAE} &= \mathcal{L}_{Denoise} + \lambda \mathcal{L}_{KL}
\end{aligned}
$$
- A denoising reconstruction loss ($\mathcal{L}_{Denoise}$) encourages the Noise Generator to produce physically plausible perturbations from which the original conformation can be recovered. This implicitly constrains the noise to respect the molecule’s underlying force fields.
- A KL divergence regularization term ($\mathcal{L}_{KL}$) pushes the generated noise distributions towards a predefined prior. This prevents the trivial solution of generating zero noise and encourages the model to explore a diverse set of low-energy conformations.
The authors also provide a theoretical analysis showing that optimizing their objective is equivalent to maximizing the Evidence Lower Bound (ELBO) on the log-likelihood of observing physically realistic conformations.
Methodology & Experimental Baselines
The model was pretrained on the PCQM4Mv2 dataset (approximately 3.4 million organic molecules) and then evaluated on a comprehensive suite of downstream tasks to test the quality of the learned representations:
- Molecular Property Prediction (QM9): The model was evaluated on 12 quantum chemical property prediction tasks for small molecules (134k molecules; 100k train, 18k val, 13k test split). DenoiseVAE achieved state-of-the-art or second-best performance on 11 of the 12 tasks, with particularly significant gains on $C_v$ (heat capacity), indicating better capture of vibrational modes.
- Force Prediction (MD17): The task was to predict atomic forces from molecular dynamics trajectories for 8 different small molecules (9,500 train, 500 val split). DenoiseVAE was the top performer on 5 of the 8 molecules (Aspirin, Benzene, Ethanol, Naphthalene, Toluene), though it underperformed Frad on Malonaldehyde, Salicylic Acid, and Uracil by significant margins.
- Ligand Binding Affinity (PDBBind v2019): On the PDBBind dataset with 30% and 60% protein sequence identity splits, the model showed strong generalization, outperforming baselines like Uni-Mol particularly on the more stringent 30% split across RMSE, Pearson correlation, and Spearman correlation.
- PCQM4Mv2 Validation: DenoiseVAE achieved a validation MAE of 0.0777 on the PCQM4Mv2 HOMO-LUMO gap prediction task with only 1.44M parameters, competitive with models 10-40x larger (e.g., GPS++ at 44.3M params achieves 0.0778).
- Ablation Studies: The authors analyzed the sensitivity to key hyperparameters, namely the prior’s standard deviation ($\sigma$) and the KL-divergence weight ($\lambda$), confirming that $\lambda=1$ and $\sigma=0.1$ are optimal. Removing the KL term leads to trivial solutions (near-zero noise). An additional ablation on the Noise Generator depth found 4 EGNN layers optimal over 2 layers. A comparison of independent (diagonal) versus non-independent (full covariance) noise sampling showed comparable results, suggesting the EGNN already captures inter-atomic dependencies implicitly.
- Case Studies: Visualizations of the learned noise variances for different molecules confirmed that the model learns chemically intuitive noise patterns. For example, it applies smaller perturbations to atoms in a rigid bicyclic norcamphor derivative and larger ones to atoms in flexible functional groups of a cyclopropane derivative. Even identical functional groups (e.g., hydroxyl) receive different noise scales in different molecular contexts.
Key Findings on Force Field Learning
- Primary Conclusion: Learning a molecule-adaptive and atom-specific noise distribution is a superior strategy for denoising-based pre-training compared to using fixed, hand-crafted heuristics. This more physically-grounded approach leads to representations that better capture molecular force fields.
- Strong Benchmark Performance: DenoiseVAE achieves best or second-best results on 11 of 12 QM9 tasks, 5 of 8 MD17 molecules, and leads on the stringent 30% LBA split. Performance is mixed on some MD17 molecules (Malonaldehyde, Salicylic Acid, Uracil), where it trails Frad.
- Effective Framework: The proposed VAE-based framework, which jointly trains a Noise Generator and a Denoising Module, is an effective and theoretically sound method for implementing this adaptive noise strategy. The interplay between the reconstruction loss and the KL-divergence regularization is key to its success.
- Limitation and Future Direction: The method is based on classical force field assumptions. The authors note that integrating more accurate force fields represents a promising direction for future work.
Reproducibility Details
Artifacts
| Artifact | Type | License | Notes |
|---|---|---|---|
| Serendipity-r/DenoiseVAE | Code | Unknown | Official implementation |
Reproducibility Status
- Source Code: The authors have released their code at Serendipity-r/DenoiseVAE on GitHub. No license is specified in the repository.
- Implementation: Hyperparameters and architectures are detailed in the paper’s appendix (A.14), and the repository provides reference implementations.
Data
- Pre-training Dataset: PCQM4Mv2 (approximately 3.4 million organic molecules)
- Property Prediction: QM9 dataset (134k molecules; 100k train, 18k val, 13k test split) for 12 quantum chemical properties
- Force Prediction: MD17 dataset (9,500 train, 500 val split) for 8 different small molecules
- Ligand Binding Affinity: PDBBind v2019 (4,463 protein-ligand complexes) with 30% and 60% sequence identity splits
Algorithms
- Noise Generator: 4-layer Equivariant Graph Neural Network (EGNN) that outputs atom-specific Gaussian noise distributions
- Denoising Module: 7-layer EGNN decoder
- Training Objective: $\mathcal{L}_{DenoiseVAE} = \mathcal{L}_{Denoise} + \lambda \mathcal{L}_{KL}$ with $\lambda=1$
- Noise Sampling: Reparameterization trick with $\tilde{x}_i = x_i + \epsilon \sigma_i$
- Prior Distribution: Standard deviation $\sigma=0.1$
Models
- Model Size: 1.44M parameters total
- Fine-tuning Protocol: Noise Generator discarded after pre-training; only the pre-trained Denoising Module (7-layer EGNN) is retained for downstream fine-tuning
- Optimizer: AdamW with cosine learning rate decay (max LR of 0.0005)
- Batch Size: 128
- System Training: Fine-tuned end-to-end for specific tasks; force prediction involves computing the gradient of the predicted energy
Evaluation
- Ablation Studies: Sensitivity analysis confirmed $\lambda=1$ and $\sigma=0.1$ as optimal hyperparameters; removing the KL term leads to trivial solutions (near-zero noise)
- Noise Generator Depth: 4 EGNN layers outperformed 2 layers across both QM9 and MD17 benchmarks
- Covariance Structure: Full covariance matrix (non-independent noise sampling) yielded comparable results to diagonal variance (independent sampling), likely because the EGNN already integrates neighboring atom information
- O(3) Invariance: The method satisfies O(3) probabilistic invariance, meaning the noise distribution is unchanged under rotations and reflections
Hardware
- GPU Configuration: Experiments conducted on a single RTX A3090 GPU; 6 GPUs with 144GB total memory sufficient for full reproduction
- CPU: Intel Xeon Gold 5318Y @ 2.10GHz
Paper Information
Citation: Liu, Y., Chen, J., Jiao, R., Li, J., Huang, W., & Su, B. (2025). DenoiseVAE: Learning Molecule-Adaptive Noise Distributions for Denoising-based 3D Molecular Pre-training. The Thirteenth International Conference on Learning Representations (ICLR).
Publication: ICLR 2025
@inproceedings{liu2025denoisevae,
title={DenoiseVAE: Learning Molecule-Adaptive Noise Distributions for Denoising-based 3D Molecular Pre-training},
author={Yurou Liu and Jiahao Chen and Rui Jiao and Jiangmeng Li and Wenbing Huang and Bing Su},
booktitle={The Thirteenth International Conference on Learning Representations},
year={2025},
url={https://openreview.net/forum?id=ym7pr83XQr}
}
Additional Resources: