Systematizing the Embedded-Atom Method
This is a Systematization (Review) paper. It consolidates the theoretical development, semi-empirical parameterization, and broad applications of the Embedded-Atom Method (EAM) into a unified framework. The paper systematizes the field by connecting the EAM to related theories (Effective Medium Theory, Finnis-Sinclair, “glue” models) and organizing phenomenological results across diverse physical regimes (bulk, surfaces, interfaces).
The authors explicitly frame the work as a survey, stating “We review here the history, development, and application of the EAM” and “This review emphasizes the physical insight that motivated the EAM.” The paper follows a classic survey structure, organizing the literature by application domains.
The Failure of Pair Potentials in Metallic Systems
The primary motivation is the failure of pair-potential models to accurately describe metallic bonding, particularly at defects and interfaces.
Physics Gap: Pair potentials assume bond strength is independent of environment, implying cohesive energy scales linearly with coordination ($Z$), whereas in reality it scales roughly as $\sqrt{Z}$.
Empirical Failures: Pair potentials incorrectly predict the “Cauchy relation” ($C_{12} = C_{44}$) and predict a vacancy formation energy equal to the cohesive energy, contradicting experimental data for fcc metals.
Practical Need: First-principles calculations (like DFT) were computationally too expensive for low-symmetry systems like grain boundaries and fracture tips, creating a need for an efficient, semi-empirical many-body potential.
Theoretical Unification & Core Innovations
The paper’s core contribution is the synthesis of the EAM as a practical computational tool that captures “coordination-dependent bond strength” without the cost of ab initio methods.
Theoretical Unification: It demonstrates that the EAM ansatz can be derived from Density Functional Theory (DFT) by assuming the total electron density is a superposition of atomic densities.
Environmental Dependence: It explicitly formulates how the “effective” pair interaction stiffens and shortens as coordination decreases (e.g., at surfaces), a feature naturally arising from the non-linearity of the embedding function.
Broad Validation: It provides a centralized evaluation of the method across a vast array of metallic properties, establishing it as the standard for atomistic simulations of face-centered cubic (fcc) metals.
Validating EAM Across Application Domains
The authors review computational experiments using Energy Minimization, Molecular Dynamics (MD), and Monte Carlo (MC) simulations across several domains:
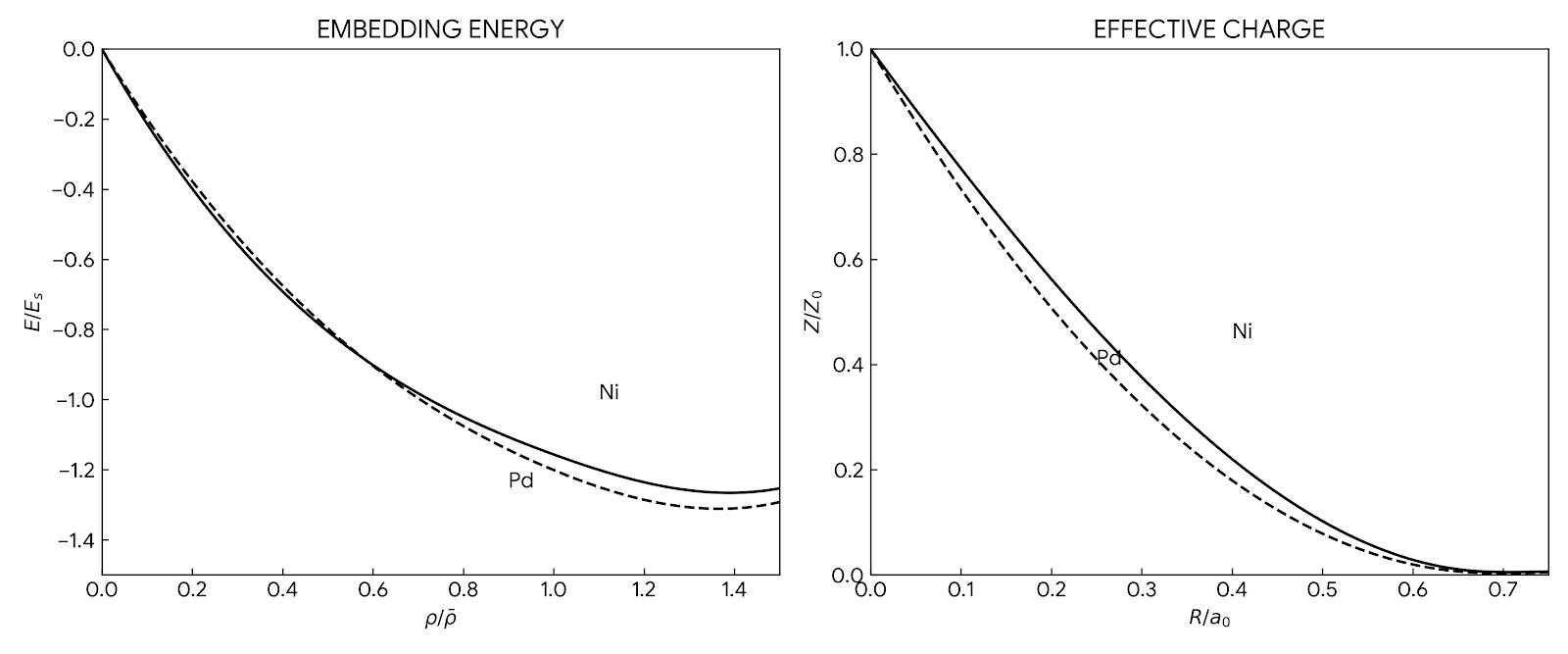
Bulk Properties: Calculation of phonon spectra, liquid structure factors, thermal expansion coefficients, and melting points for fcc metals (Ni, Pd, Pt, Cu, Ag, Au).
Defects: Computation of vacancy formation/migration energies and self-interstitial geometries.
Grain Boundaries: Calculation of grain boundary structures, energies, and elastic properties for twist and tilt boundaries in Au and Al. Computed structures show good agreement with X-ray diffraction and HRTEM experiments. The many-body interactions in the EAM produce somewhat better agreement than pair potentials, which tend to overestimate boundary expansion.
Surfaces: Analysis of surface energies, relaxations, reconstructions (e.g., Au(110) missing row), and surface phonons.
Alloys: Investigation of heat of solution, surface segregation profiles (e.g., Ni-Cu), and order-disorder transitions.
Mechanical Properties: Simulation of dislocation mobility, pinning by defects (He bubbles), and crack tip plasticity (ductile vs. brittle fracture modes).
Key Outcomes and the Limits of EAM
Many-Body Success: The EAM successfully reproduces the breakdown of the Cauchy relation and the correct ratio of vacancy formation energy to cohesive energy (~0.35) for fcc metals.
Surface Accuracy: It correctly predicts that surface bonds are shorter and stiffer than bulk bonds due to lower coordination. It accurately predicts surface reconstructions (e.g., Au(110) $(1 \times 2)$).
Alloy Behavior: The method naturally captures segregation phenomena, including oscillating concentration profiles in Ni-Cu, driven by the embedding energy.
Limitations: The method is less accurate for systems with strong directional bonding (covalent materials) or significant Fermi-surface effects, as it assumes spherically averaged electron densities.
Reproducibility Details
Data
Fitting Data: The semi-empirical functions are fitted to basic bulk properties: lattice constants, cohesive energy, elastic constants ($C_{11}$, $C_{12}$, $C_{44}$), and vacancy formation energy.
Universal Binding Curve: The cohesive energy as a function of lattice constant is constrained to follow the “universal binding curve” of Rose et al. to ensure accurate anharmonic behavior.
Alloy Data: For binary alloys, dilute heats of alloying are used for fitting cross-interactions.
Algorithms
Core Ansatz: The total energy is defined as:
$$E_{coh} = \sum_{i} G_i\left( \sum_{j \neq i} \rho_j^a(R_{ij}) \right) + \frac{1}{2} \sum_{i, j (j \neq i)} U_{ij}(R_{ij})$$
where $G$ is the embedding energy (function of local electron density $\rho$), and $U$ is a pair interaction.
Simulation Techniques:
- Molecular Dynamics (MD): Used for liquids, phonons, and fracture simulations.
- Monte Carlo (MC): Used for phase diagrams and segregation profiles (e.g., approximately $10^5$ iterations per atom).
- Phonons: Calculated via the dynamical matrix derived from the force-constant tensor $K_{ij}$.
- Normal-Mode Analysis: Vibrational normal modes obtained by diagonalizing the dynamical matrix, feasible for unit cells of up to about 260 atoms.
Models
Parameterizations: The review lists several specific function sets developed by the authors (Table 2), including:
- Daw and Baskes: For Ni, Pd, H (elemental metals and H in solution/on surfaces)
- Foiles: For Cu, Ag, Au, Ni, Pd, Pt (elemental metals)
- Foiles: For Cu, Ni (tailored for the Ni-Cu alloy system)
- Foiles, Baskes and Daw: For Cu, Ag, Au, Ni, Pd, Pt (dilute alloys)
- Daw, Baskes, Bisson and Wolfer: For Ni, H (fracture, dislocations, H embrittlement)
- Foiles and Daw: For Ni, Al (Ni-rich end of the Ni-Al alloy system)
- Daw: For Ni (calculated from first principles, not semi-empirical)
- Hoagland, Daw, Foiles and Baskes: For Al (elemental Al)
Many of these historical parameterizations are directly downloadable in machine-readable formats from the NIST Interatomic Potentials Repository (linked in the resources below).
Transferability: EAM functions are generally not transferable between different parameterization sets; mixing functions from different sets (e.g., Daw-Baskes Ni with Foiles Pd) is invalid.
Evaluation
Bulk Validation: Phonon dispersion curves for Cu show excellent agreement with experiment across the full Brillouin zone.
Thermal Properties: Linear thermal expansion coefficients match experiment well (e.g., Cu calculated: $16.4 \times 10^{-6}/K$ vs experimental: $16.7 \times 10^{-6}/K$).
Defect Energetics: Vacancy migration energies and divacancy binding energies (~0.1-0.2 eV) align with experimental data.
Surface Segregation: Correctly predicts segregation species for 18 distinct dilute alloy cases (e.g., Cu segregating in Ni).
Hardware
Compute Scale: At the time of publication (1993), Molecular Dynamics simulations of up to 35,000 atoms were possible.
Platforms: Calculations were performed on supercomputers like the CRAY-XMP, though smaller calculations were noted as feasible on high-performance workstations.
Paper Information
Citation: Daw, M. S., Foiles, S. M., & Baskes, M. I. (1993). The embedded-atom method: a review of theory and applications. Materials Science Reports, 9(7-8), 251-310. https://doi.org/10.1016/0920-2307(93)90001-U
Publication: Materials Science Reports 1993
@article{dawEmbeddedatomMethodReview1993,
title = {The embedded-atom method: a review of theory and applications},
shorttitle = {The Embedded-Atom Method},
author = {Daw, Murray S. and Foiles, Stephen M. and Baskes, Michael I.},
year = 1993,
month = mar,
journal = {Materials Science Reports},
volume = {9},
number = {7-8},
pages = {251--310},
issn = {0920-2307},
doi = {10.1016/0920-2307(93)90001-U}
}
Additional Resources: