A Prototype for Conversational Drug Compound Analysis
Method ($\Psi_{\text{Method}}$)
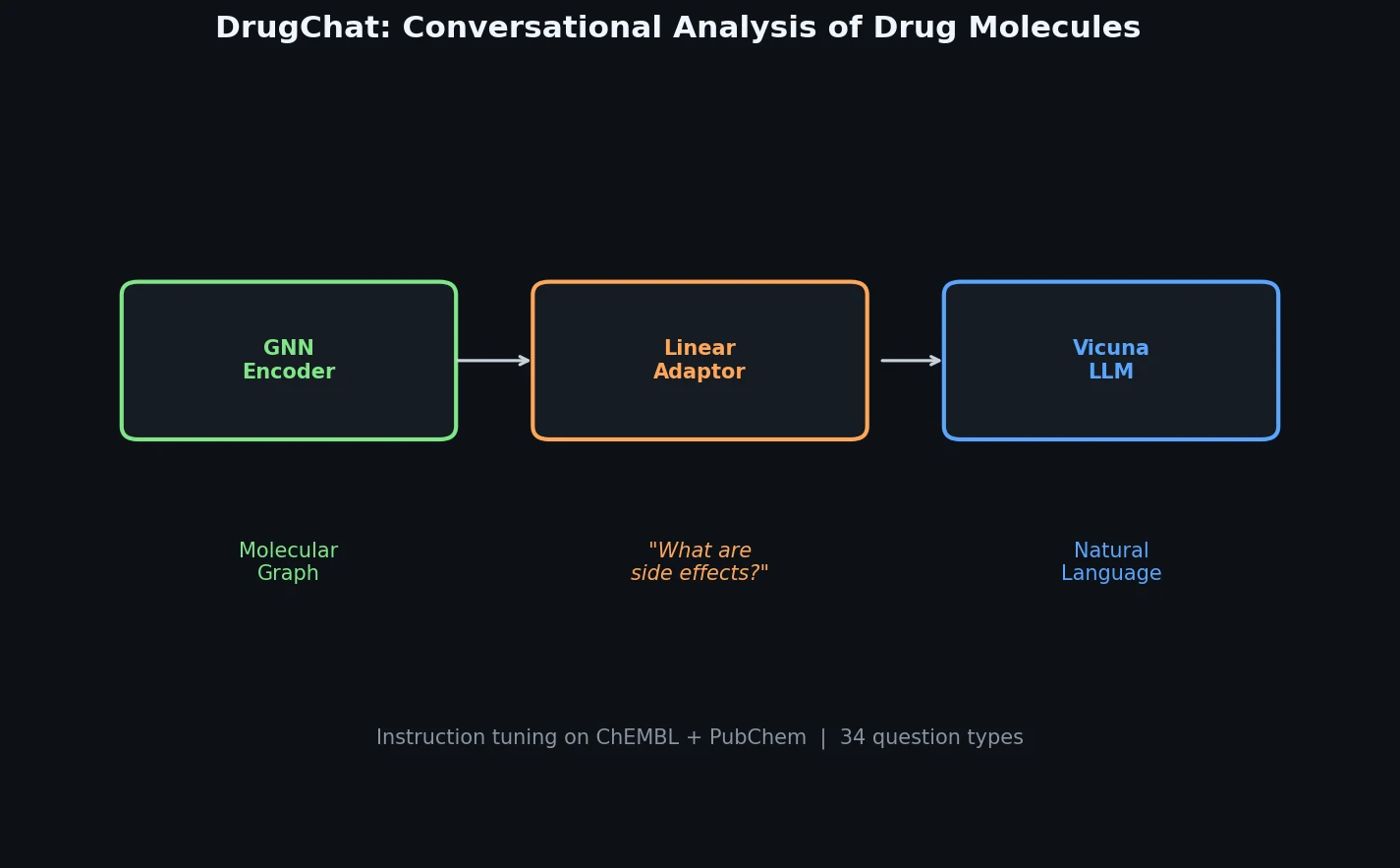
DrugChat is a prototype system that enables ChatGPT-like conversational interaction with drug molecule graphs. Users upload a compound’s molecular graph and ask free-form, multi-turn questions about its properties, mechanism of action, or therapeutic applications. The system generates natural language answers by combining a graph neural network (GNN) encoder, a large language model (LLM), and a lightweight linear adaptor that bridges the two modalities. The primary contribution is the architecture and the accompanying instruction tuning datasets (10,834 drug compounds, 143,517 QA pairs) that make this graph-to-language interaction possible.
Why Conversational Interfaces for Drug Molecules?
Drug discovery is time-intensive and expensive, often requiring years and billions of dollars to bring a single compound to market. Traditional computational chemistry tools provide specialized outputs but lack the ability to support open-ended, interactive exploration of molecular properties. Researchers working with drug compound data frequently need quick answers to diverse questions: What is the mechanism of action? Are there known drug interactions? What structural modifications could improve efficacy?
At the time of this work, large language models had demonstrated strong conversational capabilities for text, and multimodal extensions (MiniGPT-4, LLaVA) had connected vision encoders to LLMs. However, no system had bridged graph-structured molecular data with LLMs for interactive dialogue. DrugChat addresses this gap by proposing the first system (to the authors’ knowledge) that connects molecular graph representations directly to an LLM for multi-turn question answering.
Architecture: GNN-Adaptor-LLM Pipeline
The core innovation is the three-component architecture and its training strategy:
Graph Neural Network (GNN): A pre-trained GNN from Hu et al. (2020) processes the compound’s molecular graph. At each layer $k$, node representations are updated by aggregating features from neighboring nodes:
$$ h_{v}^{k} = \sigma\left(h_{v}^{k-1}, \text{AGG}\left(\left\{h_{u}^{k-1}, u \in \mathcal{N}(v)\right\}\right)\right) $$
A permutation-invariant pooling function produces the graph-level representation:
$$ h_{G} = f\left(\left\{h_{v}^{K}, v \in G\right\}\right) $$
Linear Adaptor: A single linear transformation matrix converts the GNN graph representation into a soft prompt vector compatible with the LLM’s input space. This is the only component whose weights are updated during training.
Large Language Model (Vicuna-13B): The pre-trained Vicuna-13B model takes the transformed graph prompt vector along with user questions and generates answers. Both the GNN and LLM weights remain frozen during training.
The prompt template follows the Vicuna conversational format:
$$ \mathbf{Q}: \langle\text{Graph}\rangle\langle\text{GraphFeature}\rangle\langle/\text{Graph}\rangle\langle\text{Instruction}\rangle \quad \mathbf{A}: \langle\text{Desc}\rangle $$
During training, the system minimizes a negative log-likelihood loss between generated and ground-truth answers. The entire training procedure updates only the adaptor’s parameters, making the approach computationally lightweight compared to full fine-tuning.
Instruction Tuning Datasets from ChEMBL and PubChem
The authors constructed two instruction tuning datasets:
| Dataset | Drug Compounds | QA Pairs | Source |
|---|---|---|---|
| ChEMBL | 3,892 | 129,699 | ChEMBL database (Feb 2023) |
| PubChem | 6,942 | 13,818 | PubChem (May 2023) |
| Total | 10,834 | 143,517 |
ChEMBL Dataset: Starting from 2,354,965 compounds in ChEMBL, the authors identified 14,816 with drug information and filtered to 3,892 with sufficient descriptive content. For each drug, they gathered SMILES strings, molecular features (formula, acid/base classification), and drug-specific properties (mechanism of action, therapeutic applications). They manually crafted QA pairs covering topics like rotatable bond count, Lipinski rule violations, chirality, polar surface area, development stage, approval year, and USAN classification.
PubChem Dataset: From 66,469,244 compounds in PubChem, 19,319 had drug information, and 6,942 were retained after filtering for detailed descriptions. Descriptions were sourced from ChEBI, LOTUS, and YMDB databases, yielding 13,818 QA pairs primarily asking for drug descriptions.
The QA pairs are formulaic: the ChEMBL set covers up to 34 question types per drug (an example drug in the paper shows all 34), while PubChem questions ask for descriptive summaries from different source databases.
Qualitative Demonstrations Only
The paper presents only qualitative results. Two demonstration examples show DrugChat answering multi-turn questions about test compounds not seen during training. Questions like “what makes this compound unique?” and “what diseases can this compound potentially treat?” are answered in natural language.
No systematic quantitative evaluation is reported. The authors state they “will perform a systematic quantitative evaluation by collaborating with pharmaceutical scientists,” but this evaluation is not included in the technical report.
Limitations and Future Directions
The authors identify language hallucination as the primary limitation. Since DrugChat incorporates an LLM, it may produce convincing but incorrect text descriptions about drugs, which could mislead decision-makers in real drug discovery pipelines.
Proposed mitigations include:
- Higher-quality training data and filtering strategies
- More advanced GNN encoders and LLMs
- Reinforcement learning from human feedback (RLHF) as the user base grows
Several additional limitations are worth noting:
- The QA pairs are largely factoid-style questions with short, formulaic answers, which may not capture the nuanced reasoning needed for real drug discovery tasks
- The evaluation is entirely qualitative, with no comparison to baselines or quantitative metrics
- The linear adaptor is a minimal alignment mechanism; it remains unclear how much molecular structural information is preserved through this single linear transformation
- The training data covers only a small fraction of known chemical space (10,834 compounds out of millions)
Reproducibility Details
Data
| Purpose | Dataset | Size | Notes |
|---|---|---|---|
| Training | ChEMBL Drug Instruction Tuning | 3,892 drugs, 129,699 QA pairs | From ChEMBL (Feb 2023 dump) |
| Training | PubChem Drug Instruction Tuning | 6,942 drugs, 13,818 QA pairs | From PubChem (May 2023) |
Algorithms
- GNN: Pre-trained model from Hu et al. (2020), “Strategies for Pre-training Graph Neural Networks”
- Adaptor: Single linear transformation matrix (only trainable component)
- Loss: Negative log-likelihood between generated and ground-truth answers
- Training: Only adaptor weights updated; GNN and LLM weights frozen
Models
| Component | Model | Parameters | Status |
|---|---|---|---|
| GNN Encoder | Pre-trained GNN (Hu et al., 2020) | Not specified | Frozen during training |
| LLM | Vicuna-13B | ~13B | Frozen during training |
| Adaptor | Linear projection | Not specified | Trained |
Evaluation
No quantitative evaluation metrics are reported. The paper provides only qualitative demonstrations on unseen compounds.
Hardware
No hardware specifications are reported for training or inference.
Artifacts
| Artifact | Type | License | Notes |
|---|---|---|---|
| DrugChat Code | Code | Not specified | Official implementation (repository returned 404 as of March 2026) |
Paper Information
Citation: Liang, Y., Zhang, R., Zhang, L., & Xie, P. (2023). DrugChat: Towards Enabling ChatGPT-Like Capabilities on Drug Molecule Graphs. arXiv preprint arXiv:2309.03907.
@article{liang2023drugchat,
title={DrugChat: Towards Enabling ChatGPT-Like Capabilities on Drug Molecule Graphs},
author={Liang, Youwei and Zhang, Ruiyi and Zhang, Li and Xie, Pengtao},
journal={arXiv preprint arXiv:2309.03907},
year={2023}
}