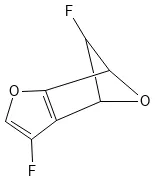
Dataset Examples

FC1C2OC1c3c(F)coc23)Related Datasets
| Dataset | Relationship | Link |
|---|---|---|
| GDB-13 | Successor | Notes |
| GDB-17 | Successor | Notes |
Key Contribution
The generation and analysis of the Generated Database (GDB), an exhaustive collection of all possible small molecules that meet specific criteria for stability and synthetic feasibility.
Overview
GDB-11 represents the first systematic enumeration of the small molecule chemical universe up to 11 atoms of C, N, O, and F. The database contains 26.4 million unique molecules corresponding to 110.9 million stereoisomers. It was created to support virtual screening and drug discovery by providing a comprehensive collection of diverse, drug-like small molecules that obey standard chemical stability rules.
Strengths
- Systematic Enumeration: Exhaustive coverage of mathematically and chemically possible structures up to 11 atoms.
- Drug-Likeness: 100% of compounds follow Lipinski’s “Rule of 5” for bioavailability, and 50% (13.2 million) follow Congreve’s more restrictive “Rule of 3” for lead-likeness.
- Structural Novelty: Features 538 newly identified ring systems that were previously unknown in existing chemical databases (like the CAS Registry or Beilstein).
- High Chirality: Over 70% of GDB molecules are chiral, providing rich 3D structural diversity, particularly in fused carbocycles and heterocycles.
Limitations
- Size Restriction: Strictly limited to small molecules with a maximum of 11 heavy atoms.
- Element Restriction: Only contains C, N, O, and F. Important biological and pharmaceutical elements like Phosphorus (P), Sulfur (S), and Silicon (Si) are excluded to prevent combinatorial explosion.
- Excluded Topologies: Excludes highly strained molecules (e.g., specific bridged systems), allenes, and bridgehead double bonds.
- Unstable Functional Groups: Excludes chemical classes deemed unstable or highly reactive (e.g., gem-diols, hemiacetals, aminals, enols, orthoacids).
- Computational Nature: Consists entirely of computer-generated, theoretical structures without experimental synthesis or biological validation.
Technical Notes
Construction
Graph Selection
The program GENG was used to generate an initial set of 843,335 connected graphs with up to 11 nodes and a maximum node connectivity of 4. These were filtered to 15,726 stable saturated hydrocarbon graphs using:
- Topological Criteria: Removed graphs with a node in multiple small (3- or 4-membered) rings, tetravalent bridgeheads in small rings, and nonplanar graphs (e.g., Claus-benzol).
- Steric Criteria: Graphs containing highly distorted centers were removed using an adapted MM2 force field energy-minimization with a cutoff of +17 kcal/mol.
Structure Generation
Graph symmetry algorithms identified valid locations for unsaturations and heteroatoms (C, N, O, F). Specific valence constraints were continuously enforced. Combinatorial distribution of elements and multiple bonds (excluding bridgehead double bonds, triple bonds in rings smaller than nine, and allenes) yielded a theoretical “dark matter universe” (DMU) of over 1.7 billion unique structures.
Filters
The 1.7 billion structural candidates contained unstable environments which were aggressively filtered, reducing the set to 27.7 million possible stable molecules. Rejected unstable/reactive features included:
- High-Energy Bonds: Gem-diols, non-stabilized aminals, hemiaminals, enols, orthoesters, unstable imines, acyl fluorides, and geminal di-heteroatoms.
- Heteroatom-Heteroatom Bonds: Peroxides (O-O), N-O, N-N, N-F, and triazanes, unless stabilized (e.g., hydrazones, oximes).
- Strained Topologies: 3/4-membered rings containing N-N or N-O bonds, and bridgehead heteroatom bonds causing instabilities (like Bredt’s rule violations).
Removal of redundant tautomeric forms collapsed the set to the foundational 26.4 million structures.
Stereoisomer Generation
Stereoisomers were cleanly enumerated by identifying all asymmetric centers and functional double bonds, blocking Z/E isomerism in rings smaller than 10 nodes. From the 26.4 million unique constitutional isomers, 110.9 million stereoisomers were generated (averaging 4.2 stereoisomers per molecule).
Analysis Methodology
Kohonen Maps (Self-Organizing Maps)
The chemical space visualization and compound class analysis used a Kohonen Map (Self-Organizing Map/SOM):
- Input Features: 48-dimensional autocorrelation vectors encoding topological relationships and atomic properties. The autocorrelation vector $\text{AC}_d$ for a topological distance $d$ is defined as:
$$ \text{AC}_d = \sum_{i=1}^{N} \sum_{j=1}^{N} \delta (p_i p_j)_d $$
(where $N$ is the number of atoms, $p$ is the atomic property, and $\delta (p_i, p_j)_d = p_i p_j$ if the topological distance between atoms $i$ and $j$ is $d$, and 0 otherwise).
- Training Data: Random subset of 1,000,000 GDB molecules
- Architecture: 200x200 neuron grid
- Training Protocol: 250,000 epochs with 100 molecules presented per epoch
- Algorithm: Standard Kohonen algorithm
- Key Insight: Reveals that “lead-like” compounds cluster in chiral regions of fused carbocycles/heterocycles
Comparison
The full database was compared comprehensively to a Reference Database (RDB) of 63,857 known compounds (up to 11 atoms) extracted from PubChem, ChemACX, ChemSCX, NCI Open Database, and the Merck Index. Of the 63,857 RDB compounds, 37,393 (58.6%) were found in GDB. The remaining 26,464 compounds were absent due to structural rule violations, exclusion of elements beyond C/N/O/F, and filtered unstable chemistries.
New Rings
All 309 entirely acyclic graphs in GDB mapped cleanly to published structures. External databases contained only 670 of the 1,208 purely cyclic theoretical ring systems (55.5%). Furthermore, 367 of the 538 newly identified ring systems (68.2%) express inherently chiral topologies.
Stereochemistry
Small molecules under 5 heavy atoms skew strongly towards simple achiral structures. As the atom count increases, a dominant stereochemical shift emerges: over two-thirds of structures containing exactly 10 or 11 atoms occupy chiral configuration spaces. Approximately 86% of the molecules in GDB contain exactly 11 atoms (22.8 million of 26.4 million).
Physicochemical Properties
Because all GDB molecules contain at most 11 heavy atoms, 100% of them satisfy Lipinski’s “Rule of 5” for bioavailability. Under the more restrictive Congreve “Rule of 3” for lead-likeness (MW < 300, RBC < 3, logP < 3, HBDC < 3, HBAC < 3, TPSA < 60 $\text{\AA}^2$), exactly 50% (13.2 million structures) qualify. Virtual screening using the Molinspiration miscreen toolkit (Bayesian statistics-based) identified 42,804 virtual hits across three drug target classes: 3,043 kinase inhibitor candidates, 24,489 GPCR ligand candidates, and 19,696 ion-channel modulator candidates. Of these virtual hits, 59.8% occupied Kohonen map neurons not populated by any known RDB compound.
Reproducibility Details
While the generated GDB-11 database is openly available, reproducing the exact generation from graph to stereoisomer relies on in-house and proprietary software which is not publicly available.
| Artifact | Type | License | Notes |
|---|---|---|---|
| GDB Downloads (University of Berne) | Dataset | Unknown | Official host for GDB databases |
| Zenodo Record (10.5281/zenodo.5172017) | Dataset | Unknown | Version-agnostic Zenodo archive of GDB-11 |
- Paper Accessibility: Closed-access (Published in JCIM 2007; no preprint available).
- Data Availability: The complete dataset is hosted on an open-access Zenodo repository (version-agnostic DOI): 10.5281/zenodo.5172017.
- Software Dependencies (Closed/Commercial):
- Generation code is a closed-source Java (J2SE v5.0) application.
- Relies on proprietary ChemAxon libraries (JChem v3.1, Marvin v4.0 API).
- Virtual screening evaluation utilized the commercial Molinspiration
miscreentoolkit.
- Hardware Profile:
- CPUs: Two AMD Opteron 252 2.6 GHz processors
- Parallelization: 80-fold parallelization
- Compute Time: Approximately 20 hours for full generation
Force Field
A custom implementation of the MM2 force field was used for steric energy minimization during structure validation. It used the parameter set from Allinger, specifically adding a quartic term for bond stretching to prevent bond lengthening far from equilibrium:
$$ \begin{aligned} E_{\text{Steric}} &= \sum_{\text{bonds}} k_b(l_i - l_{0,i})^2 \left[1 + k’_b(l_i - l_{0,i}) + k’’_b(l_i - l_{0,i})^2\right] \\ &\quad + \sum_{\text{angles}} k_\theta(\theta_i - \theta_{0,i})^2 \left[1 + k’_\theta(\theta_i - \theta_{0,i})^4\right] \\ &\quad + \sum_{\text{angles}} k_{b,\theta}(\theta_i - \theta_{0,i})^2 \left[(l_a - l_{0,a}) + (l_b - l_{0,b})\right] \\ &\quad + \sum_{\text{torsions}} \left[ \frac{V_1}{2} (1 + \cos \omega) + \frac{V_2}{2} (1 - \cos 2\omega) + \frac{V_3}{2} (1 + \cos 3\omega) \right] \\ &\quad + \sum_{i=1}^N \sum_{j=i+1}^N \epsilon_{ij} \left[ A \exp \left( \frac{-B r_{ij}}{\sum r^{\ast}_{ij}} \right) - C \left( \frac{r_{ij}}{\sum r^{\ast}_{ij}} \right)^6 \right] \end{aligned} $$
Citation
@article{fink2007virtual,
title={Virtual exploration of the chemical universe up to 11 atoms of C, N, O, and F: assembly of 26.4 million structures (110.9 million stereoisomers) and analysis for new ring systems, stereochemistry, physicochemical properties, compound classes, and drug discovery},
author={Fink, Tobias and Reymond, Jean-Louis},
journal={Journal of Chemical Information and Modeling},
volume={47},
number={2},
pages={342-353},
year={2007},
publisher={ACS Publications}
}