ZINC-22: A Multi-Billion Scale Database for Ligand Discovery
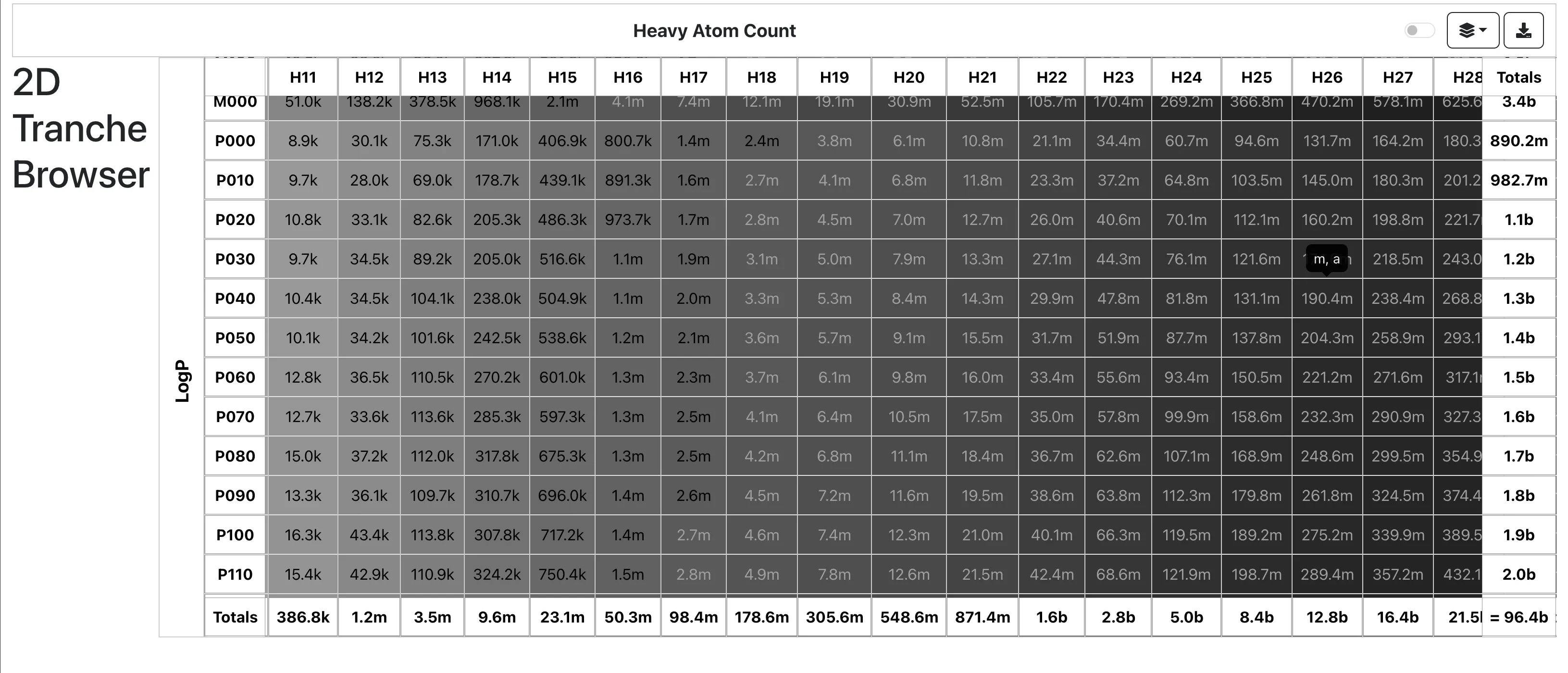
ZINC-22 is a multi-billion-scale public database containing over 37 billion make-on-demand molecules. It utilizes distributed infrastructure and specialized search algorithms to support modern ultra-large virtual screening campaigns.