A Systematization of Transformers for Molecular Property Prediction
This is a Systematization paper. Sultan et al. provide the first comprehensive, structured review of sequence-based transformer models applied to molecular property prediction (MPP). The review catalogs 16 models published between 2019 and 2023, organizes them by architecture type (encoder-decoder, encoder-only, decoder-only), and systematically examines seven key design decisions that arise when building a transformer for MPP. The paper’s primary contribution is identifying gaps in current evaluation practices and articulating what standardization the field needs for meaningful progress.
The Problem: Inconsistent Evaluation Hinders Progress
Molecular property prediction is essential for drug discovery, crop protection, and environmental science. Deep learning approaches, including transformers, have been increasingly applied to this task by learning molecular representations from string notations like SMILES and SELFIES. However, the field faces several challenges:
- Small labeled datasets: Labeled molecular property datasets typically contain only hundreds or thousands of molecules, making supervised learning alone insufficient.
- No standardized evaluation protocol: Different papers use different data splits (scaffold vs. random), different splitting implementations, different numbers of repetitions (3 to 50), and sometimes do not share their test sets. This makes direct comparison across models infeasible.
- Unclear design choices: With many possible configurations for pre-training data, chemical language, tokenization, positional embeddings, model size, pre-training objectives, and fine-tuning approaches, the field lacks systematic analyses to guide practitioners.
The authors note that standard machine learning methods with fixed-size molecular fingerprints remain strong baselines for real-world datasets, illustrating that the promise of transformers for MPP has not yet been fully realized.
Seven Design Questions for Molecular Transformers
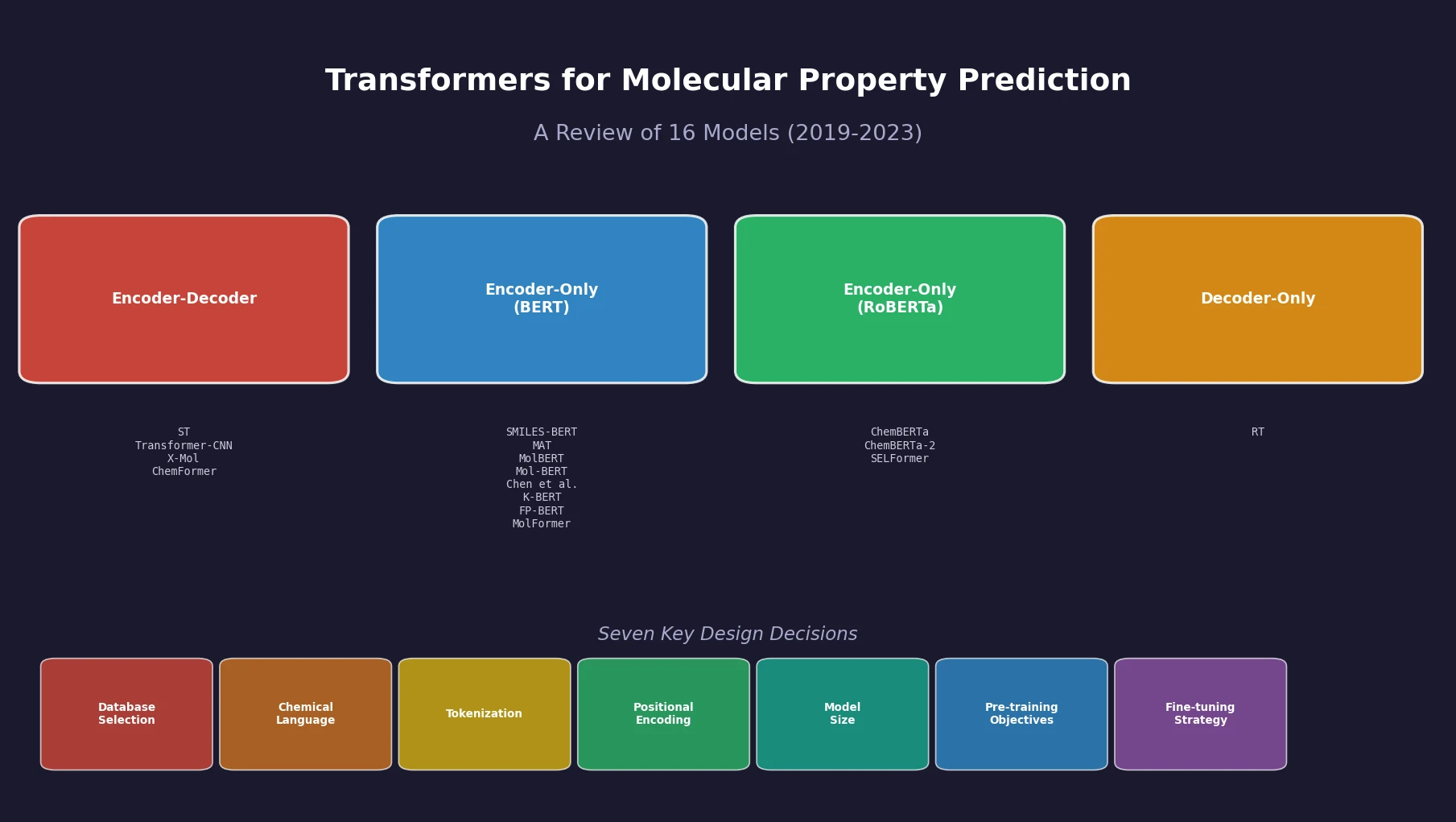
The central organizing framework of this review addresses seven questions practitioners must answer when building a transformer for MPP. For each, the authors synthesize findings across the 16 reviewed models.
Reviewed Models
The paper catalogs 16 models organized by architecture:
| Architecture | Base Model | Models |
|---|---|---|
| Encoder-Decoder | Transformer, BART | ST, Transformer-CNN, X-Mol, ChemFormer |
| Encoder-Only | BERT | SMILES-BERT, MAT, MolBERT, Mol-BERT, Chen et al., K-BERT, FP-BERT, MolFormer |
| Encoder-Only | RoBERTa | ChemBERTa, ChemBERTa-2, SELFormer |
| Decoder-Only | XLNet | Regression Transformer (RT) |
The core attention mechanism shared by all these models is the scaled dot-product attention:
$$ \text{Attention}(Q, K, V) = \text{softmax}\left(\frac{QK^{T}}{\sqrt{d_{k}}}\right)V $$
where $Q$, $K$, and $V$ are the query, key, and value matrices, and $d_{k}$ is the dimension of the key vectors.
Question 1: Which Database and How Many Molecules?
Pre-training data sources vary considerably. The three main databases are ZINC (37 billion molecules in ZINC22), ChEMBL (2.4 million unique molecules with 20 million bioactivity measurements), and PubChem (111 million unique molecules). Pre-training set sizes ranged from 900K (ST on ChEMBL) to 1.1B molecules (MolFormer on ZINC + PubChem).
| Model | Database | Size | Language |
|---|---|---|---|
| ST | ChEMBL | 900K | SMILES |
| MolBERT | ChEMBL (GuacaMol) | 1.6M | SMILES |
| ChemBERTa | PubChem | 100K-10M | SMILES, SELFIES |
| ChemBERTa-2 | PubChem | 5M-77M | SMILES |
| MAT | ZINC | 2M | List of atoms |
| MolFormer | ZINC + PubChem | 1.1B | SMILES |
| Chen et al. | C, CP, CPZ | 2M-775M | SMILES |
A key finding is that larger pre-training datasets do not consistently improve downstream performance. MolFormer showed minimal difference between models trained on 100M vs. 1.1B molecules. ChemBERTa-2 found that the model trained on 5M molecules using MLM performed comparably to 77M molecules for BBBP (both around 0.70 ROC-AUC). Chen et al. reported comparable $R^{2}$ values of $0.925 \pm 0.01$, $0.917 \pm 0.012$, and $0.915 \pm 0.01$ for ESOL across datasets of 2M, 103M, and 775M molecules, respectively. The data composition and covered chemical space appear to matter more than raw size.
Question 2: Which Chemical Language?
Most models use SMILES. ChemBERTa, RT, and SELFormer also explored SELFIES. MAT uses a simple list of atoms with structural features, while Mol-BERT and FP-BERT use circular fingerprints.
Direct comparisons between SMILES and SELFIES (by ChemBERTa on Tox21 SR-p53 and RT for drug-likeness prediction) found no significant performance difference. The RT authors reported that SELFIES models performed approximately $0.004 \pm 0.01$ better on RMSE, while SMILES models performed approximately $0.004 \pm 0.01$ better on Pearson correlation. The choice of chemical language does not appear to be a major factor in prediction performance, and even non-string representations (atom lists in MAT, fingerprints in Mol-BERT) perform competitively.
Question 3: How to Tokenize?
Tokenization methods span atom-level (42-66 vocabulary tokens), regex-based (47-2,362 tokens), BPE (509-52K tokens), and substructure-based (3,357-13,325 tokens) approaches. No systematic comparison of tokenization strategies exists in the literature. The vocabulary size varied dramatically, from 42 tokens for MolBERT to over 52K for ChemBERTa. The authors argue that chemically meaningful tokenization (e.g., functional group-based fragmentation) could improve both performance and explainability.
Question 4: How to Add Positional Embeddings?
Most models inherited the absolute positional embedding from their NLP base models. MolBERT and RT adopted relative positional embeddings. MolFormer combined absolute and Rotary Positional Embedding (RoPE). MAT incorporated spatial information (inter-atomic 3D distances and adjacency) alongside self-attention.
MolFormer’s comparison showed that RoPE became superior to absolute embeddings only when the pre-training dataset was very large. The performance difference (MAE on QM9) between absolute and RoPE embeddings for models trained on 111K, 111M, and 1.1B molecules was approximately $-0.20 \pm 0.18$, $-0.44 \pm 0.22$, and $0.27 \pm 0.12$, respectively.
The authors highlight that SMILES and SELFIES are linearizations of a 2D molecular graph, so consecutive tokens in a sequence are not necessarily spatially close. Positional embeddings that reflect 2D or 3D molecular structure remain underexplored.
Question 5: How Many Parameters?
Model sizes range from approximately 7M (ST, Mol-BERT) to over 100M parameters (MAT). Most chemical language models operate with 100M parameters or fewer, much smaller than NLP models like BERT (110M-330M) or GPT-3 (175B).
| Model | Dimensions | Heads | Layers | Parameters |
|---|---|---|---|---|
| ST | 256 | 4 | 4 | 7M |
| MolBERT | 768 | 12 | 12 | 85M |
| MolFormer | 768 | 12 | 6, 12 | 43M, 85M |
| SELFormer | 768 | 12, 4 | 8, 12 | 57M, 85M |
| MAT | 1024 | 16 | 8 | 101M |
| ChemBERTa | 768 | 12 | 6 | 43M |
SELFormer and MolFormer both tested different model sizes. SELFormer’s larger model (approximately 86M parameters) showed approximately 0.034 better ROC-AUC for BBBP compared to the smaller model. MolFormer’s larger model (approximately 87M parameters) performed approximately 0.04 better ROC-AUC on average for BBBP, HIV, BACE, and SIDER. The field lacks the systematic scaling analyses (analogous to Kaplan et al. and Hoffmann et al. in NLP) needed to establish proper scaling laws for chemical language models.
Question 6: Which Pre-training Objectives?
Pre-training objectives fall into domain-agnostic and domain-specific categories:
| Model | Pre-training Objective | Fine-tuning |
|---|---|---|
| MolFormer | MLM | Frozen, Update |
| SMILES-BERT | MLM | Update |
| MolBERT | MLM, PhysChemPred, SMILES-EQ | Frozen, Update |
| K-BERT | Atom feature, MACCS prediction, CL | Update last layer |
| ChemBERTa-2 | MLM, MTR | Update |
| MAT | MLM, 2D Adjacency, 3D Distance | Update |
| ChemFormer | Denoising Span MLM, Augmentation | Update |
| RT | PLM (Permutation Language Modeling) | - |
Domain-specific objectives (predicting physico-chemical properties, atom features, or MACCS keys) showed promising but inconsistent results. MolBERT’s PhysChemPred performed closely to the full three-objective model (approximately $0.72 \pm 0.06$ vs. $0.71 \pm 0.06$ ROC-AUC in virtual screening). The SMILES-EQ objective (identifying equivalent SMILES) was found to lower performance when combined with other objectives. K-BERT’s contrastive learning objective did not significantly change performance (average ROC-AUC of 0.806 vs. 0.807 with and without CL).
ChemBERTa-2’s Multi-Task Regression (MTR) objective performed noticeably better than MLM-only for almost all four classification tasks across pre-training dataset sizes.
Question 7: How to Fine-tune?
Fine-tuning through weight updates generally outperforms frozen representations. SELFormer showed this most dramatically, with a difference of 2.187 RMSE between frozen and updated models on FreeSolv. MolBERT showed a much smaller difference (0.575 RMSE on FreeSolv), likely because its domain-specific pre-training objectives already produced representations closer to the downstream tasks.
Benchmarking Challenges and Performance Comparison
Downstream Datasets
The review focuses on nine benchmark datasets across three categories from MoleculeNet:
| Dataset | Molecules | Tasks | Type | Application |
|---|---|---|---|---|
| ESOL | 1,128 | 1 regression | Physical chemistry | Aqueous solubility |
| FreeSolv | 642 | 1 regression | Physical chemistry | Hydration free energy |
| Lipophilicity | 4,200 | 1 regression | Physical chemistry | LogD at pH 7.4 |
| BBBP | 2,050 | 1 classification | Physiology | Blood-brain barrier |
| ClinTox | 1,484 | 2 classification | Physiology | Clinical trial toxicity |
| SIDER | 1,427 | 27 classification | Physiology | Drug side effects |
| Tox21 | 7,831 | 12 classification | Physiology | Nuclear receptor/stress pathways |
| BACE | 1,513 | 1 classification | Biophysics | Beta-secretase 1 binding |
| HIV | 41,127 | 1 classification | Biophysics | Anti-HIV activity |
Inconsistencies in Evaluation
The authors document substantial inconsistencies that prevent fair model comparison:
- Data splitting: Models used different splitting methods (scaffold vs. random) and different implementations even when using the same method. Not all models adhered to scaffold splitting for classification tasks as recommended.
- Different test sets: Even models using the same split type may not evaluate on identical test molecules due to different random seeds.
- Varying repetitions: Repetitions ranged from 3 (RT) to 50 (Chen et al.), making some analyses more statistically robust than others.
- Metric inconsistency: Most use ROC-AUC for classification and RMSE for regression, but some models report only averages without standard deviations, while others report standard errors.
Performance Findings
When comparing only models evaluated on the same test sets (Figure 2 in the paper), the authors observe that transformer models show comparable, but not consistently superior, performance to existing ML and DL models. The performance varies considerably across models and datasets.
For BBBP, the Mol-BERT model reported lower ROC-AUC than its corresponding MPNN (approximately 0.88 vs. 0.91), while MolBERT outperformed its corresponding CDDD model (approximately 0.86 vs. 0.76 ROC-AUC) and its SVM baseline (approximately 0.86 vs. 0.70 ROC-AUC). A similar mixed pattern appeared for HIV: ChemBERTa performed worse than its corresponding ML models, while MolBERT performed better than its ML (approximately 0.08 higher ROC-AUC) and DL (approximately 0.03 higher ROC-AUC) baselines. For SIDER, Mol-BERT performed approximately 0.1 better ROC-AUC than its corresponding MPNN. For regression, MAT and MolBERT showed improved performance over their ML and DL baselines on ESOL, FreeSolv, and Lipophilicity. For example, MAT performed approximately 0.2 lower RMSE than an SVM model and approximately 0.03 lower RMSE than the Weave model on ESOL.
Key Takeaways and Future Directions
The review concludes with six main takeaways:
- Performance: Transformers using SMILES show comparable but not consistently superior performance to existing ML and DL models for MPP.
- Scaling: No systematic analysis of model parameter scaling relative to data size exists for chemical language models. Such analysis is essential.
- Pre-training data: Dataset size alone is not the sole determinant of downstream performance. Composition and chemical space coverage matter.
- Chemical language: SMILES and SELFIES perform similarly. Alternative representations (atom lists, fingerprints) also work when the architecture is adjusted.
- Domain knowledge: Domain-specific pre-training objectives show promise, but tokenization and positional encoding remain underexplored.
- Benchmarking: The community needs standardized data splitting, fixed test sets, statistical analysis, and consistent reporting to enable meaningful comparison.
The authors also highlight the need for attention visualization and explainability analysis, investigation of NLP-originated techniques (pre-training regimes, fine-tuning strategies like LoRA, explainability methods), and adaptation of these techniques to the specific characteristics of chemical data (smaller vocabularies, shorter sequences).
Reproducibility Details
Data
This is a review paper. No new data or models are introduced. All analyses use previously reported results from the 16 reviewed papers, with additional visualization and comparison. The authors provide a GitHub repository with the code and data used to generate their comparative figures.
Algorithms
Not applicable (review paper). The paper describes training strategies at a conceptual level, referencing the original publications for implementation details.
Models
Not applicable (review paper). The paper catalogs 16 models with their architecture details, parameter counts, and training configurations across Tables 1, 4, 5, 6, and 7.
Evaluation
The paper compiles performance across nine MoleculeNet datasets. Key comparison figures (Figures 2 and 7) restrict to models evaluated on the same test sets for fair comparison, using ROC-AUC for classification and RMSE for regression.
Hardware
Not applicable (review paper).
| Artifact | Type | License | Notes |
|---|---|---|---|
| Transformers4MPP_review | Code | MIT | Figure generation code and compiled data |
Paper Information
Citation: Sultan, A., Sieg, J., Mathea, M., & Volkamer, A. (2024). Transformers for Molecular Property Prediction: Lessons Learned from the Past Five Years. Journal of Chemical Information and Modeling, 64(16), 6259-6280. https://doi.org/10.1021/acs.jcim.4c00747
@article{sultan2024transformers,
title={Transformers for Molecular Property Prediction: Lessons Learned from the Past Five Years},
author={Sultan, Afnan and Sieg, Jochen and Mathea, Miriam and Volkamer, Andrea},
journal={Journal of Chemical Information and Modeling},
volume={64},
number={16},
pages={6259--6280},
year={2024},
publisher={American Chemical Society},
doi={10.1021/acs.jcim.4c00747}
}