A Systematization of Chemical Language Models for Molecular Generation
This paper is a Systematization that provides a comprehensive, PRISMA-guided systematic review of deep learning chemical language models (CLMs) used for de novo molecular generation. The primary contribution is a structured statistical analysis of 72 retrieved articles from 2020 to June 2024, comparing architectures (RNNs, transformers, VAEs, GANs, S4 models), molecular representations, biased generation strategies, and quality metrics from the MOSES and GuacaMol benchmarking platforms. The review addresses five research questions about architecture configuration effects, best-performing architectures, impactful hyperparameters, common molecular representations, and effective biased generation methods.
Motivation: Evaluating Four Years of Generative CLM Progress
Deep learning molecular generation has expanded rapidly since 2018, when Gomez-Bombarelli et al. and Segler et al. demonstrated that deep generative models could learn to produce novel molecules from SMILES representations. By 2020, multiple architectures (RNNs, transformers, VAEs, GANs) were being applied to chemical language modeling, and benchmarking platforms like MOSES and GuacaMol had been introduced to enable standardized evaluation.
Despite this growth, existing reviews largely focused on theoretical background or drug development applications rather than systematic statistical comparison of model performance. Few studies had examined how architecture choice, training dataset size, molecular representation format, and biased learning strategies interact to affect generation quality metrics like validity, uniqueness, and novelty. This review fills that gap by restricting the analysis to papers reporting MOSES or GuacaMol metrics, enabling quantitative cross-study comparison.
PRISMA-Based Systematic Review Methodology
The review follows the Preferred Reporting Items for Systematic Review and Meta-Analysis (PRISMA) guidelines. Articles were retrieved from Scopus, Web of Science, and Google Scholar using six Boolean search queries combining terms like “Molecule Generation,” “Chemical Language Models,” “Deep Learning,” and specific architecture names. The search window covered January 2020 to June 2024.
Eligibility Criteria
Papers were included if they:
- Were written in English
- Explicitly presented at least two metrics of uniqueness, validity, or novelty
- Defined these metrics consistent with MOSES or GuacaMol concepts
- Used deep learning generative models for de novo molecule design
- Used conventional (non-quantum) deep learning methods
- Were published between January 2020 and June 2024
This yielded 48 articles from query-based search and 25 from citation search, totaling 72 articles. Of these, 62 used CLM approaches (string-based molecular representations) and 10 used graph-based representations.
Data Collection
For each article, the authors extracted: journal details, database name, training dataset size, molecular representation type (SMILES, SELFIES, InChI, DeepSMILES), architecture details (embedding length, layers, hidden units, trainable parameters, dropout, temperature, batch size, epochs, learning rate, optimizer), biased method usage (TL, RL, conditional learning), and generation metrics (validity, uniqueness, novelty, scaffold diversity, SNN, FCD).
Evaluation Metrics
The review focuses on three core MOSES metrics:
$$ \text{Validity}(V_m) = \frac{\text{Valid molecules}}{\text{Molecules produced}} $$
$$ \text{Uniqueness} = \frac{\text{set}(V_m)}{V_m} $$
$$ \text{Novelty} = 1 - \frac{V_m \cap T_d}{V_m} $$
where $V_m$ denotes valid molecules and $T_d$ the training dataset.
Architecture Distribution and Performance Comparison
Architecture Trends (2020-2024)
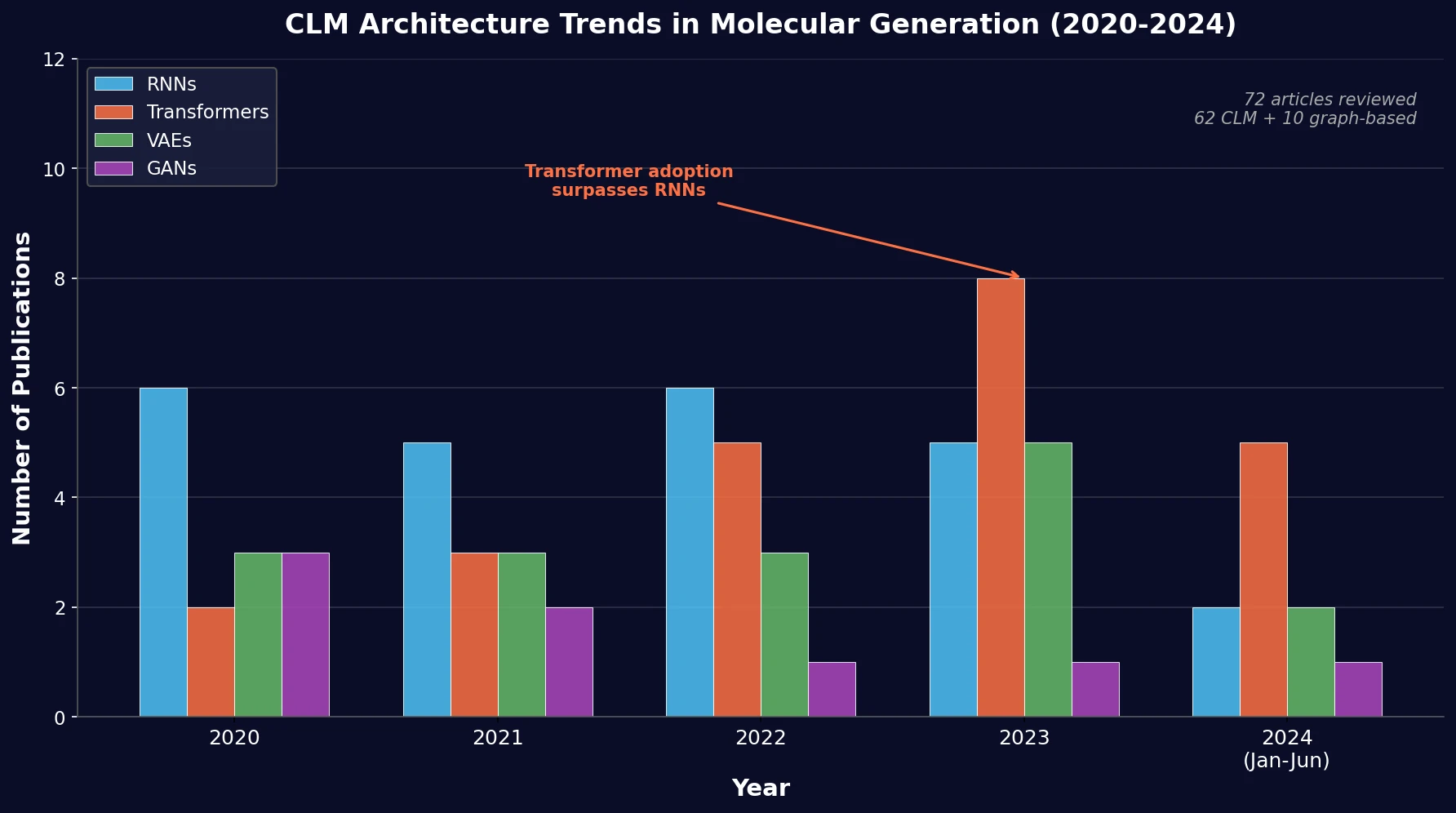
The review found that RNNs and transformers dominate CLM usage, with a growing trend toward transformers over time. The breakdown across 62 CLM articles: 24 RNN-based, 23 transformer-based, 16 VAE-based, 8 GAN-based, and 1 S4-based model. Among RNN variants, LSTM was the most common, followed by GRU, despite GRU having fewer trainable parameters.
The increase in transformer adoption is attributed to self-attention mechanisms enabling parallel computation and effective long-range dependency capture. Meanwhile, GANs and VAEs saw lower adoption rates, partly due to higher memory and time complexity and reduced ability to generate large molecules.
Molecular Representations and Databases
SMILES was used exclusively in 77.27% of CLM articles, reflecting its wide database availability and compact format. SELFIES, DeepSMILES, and InChI each appeared in smaller fractions. The dominant databases were ChEMBL and ZINC (27 articles each), followed by PubChem (4 articles). Approximately 71% of reviewed articles focused on drug discovery applications.
| Database | Molecules (millions) | Representation | Articles |
|---|---|---|---|
| ChEMBL | 2.4 | SMILES, InChI | 27 |
| ZINC | 750 | SMILES | 27 |
| PubChem | 115.3 | SMILES, InChI | 4 |
| COCONUT | 0.695 | SMILES, InChI | 1 |
| DNA-Encoded Library | 1,040 | SMILES | 1 |
Unbiased Model Performance
Validity: No statistically significant differences were observed across architecture families. Transformers generally achieved high validity through self-attention mechanisms that retain uncompressed sequence information. However, one transformer model (TransMol) achieved only 6.9% validity when using stochastic sampling with Gaussian noise to explore unseen chemical space. GANs showed high dispersion, with validity as low as 8.5% when learning from gene expression signatures rather than molecular structures directly.
Uniqueness: No significant differences in median uniqueness across architectures. Transformer-based models using masked self-attention achieved near-perfect uniqueness scores. Scaffold decoration and fragment-linking approaches sometimes compromised uniqueness due to overfit-driven redundancy.
Validity-Novelty Trade-off: The authors propose a “Valid/Sample” metric (Validity x Novelty) and find an inverse trend between validity and novelty (Spearman $\rho = -0.3575$, p-value = 0.0618). Only 17.9% of models achieved above-median values for both validity (95.6%) and novelty (96.5%) simultaneously. SELFIES-based models achieve 100% validity by construction, which can help address this trade-off.
Biased Model Performance
The review examines three biased generation strategies:
Transfer Learning (TL): The most prevalent biased method, used across all architecture types. Fine-tuning transfers pre-trained parameters to a target model, requiring significantly fewer training molecules (median ~2,507 vs. ~1.1M for unbiased). TL does not significantly affect validity (p = 0.16) or novelty (p = 0.84), but uniqueness decreases significantly (median 90.2% vs. 97.9%, p = 0.014), likely due to overfitting on small target datasets.
| Metric | Unbiased (median) | TL Target (median) | p-value |
|---|---|---|---|
| Training size | 1,128,920 | 2,507 | <0.0001 |
| Validity | 98.05% | 95.5% | 0.1602 |
| Uniqueness | 97.9% | 90.2% | 0.0144 |
| Novelty | 91.6% | 96.0% | 0.8438 |
Reinforcement Learning (RL): Applied only to RNNs and transformers in the reviewed set. 90.1% of RL implementations used policy gradient methods with scoring functions for properties like synthesizability, binding affinity, and membrane permeability. No significant effects on generation metrics were observed.
| Metric | Unbiased (median) | RL Target (median) | p-value |
|---|---|---|---|
| Validity | 91.1% | 96.5% | 0.1289 |
| Uniqueness | 99.9% | 89.7% | 0.0935 |
| Novelty | 91.5% | 93.5% | 0.2500 |
Conditional Learning (CL): Integrates domain-specific data (properties, bioactivities, functional groups) directly into training via constraint tokens or property embeddings. Used primarily with encoder-decoder architectures (ARAEs, VAEs, transformers). CL does not significantly degrade generation metrics relative to unbiased models.
| Metric | Unbiased (median) | CL Target (median) | p-value |
|---|---|---|---|
| Validity | 98.5% | 96.8% | 0.4648 |
| Uniqueness | 99.9% | 97.5% | 0.0753 |
| Novelty | 89.3% | 99.6% | 0.2945 |
Key Findings and Directions for Chemical Language Models
Main Conclusions
Transformers are overtaking RNNs as the dominant CLM architecture, driven by self-attention mechanisms that capture long-range dependencies without the gradient vanishing issues of recurrent models.
SMILES remains dominant (77% of models) despite known limitations (non-uniqueness, syntax errors). SELFIES shows promise for improving the validity-novelty trade-off.
No architecture achieves both high validity and high novelty easily. Only 17.9% of unbiased models exceeded medians for both metrics simultaneously, highlighting a fundamental tension in generative chemistry.
Transfer learning requires only ~2,500 molecules to generate targeted compounds, compared to ~1.1M for unbiased training, but at the cost of reduced uniqueness.
Combining biased methods (e.g., TL + RL, CL + TL) shows promise for multi-objective optimization and exploring distant regions of chemical space.
S4 models were newly introduced for CLMs in 2023, showing competitive performance with the dual nature of convolution during training and recurrent generation.
Limitations
The review is restricted to papers reporting MOSES or GuacaMol metrics, which excludes many molecular generation studies that use alternative evaluation frameworks. The statistical comparisons rely on median values reported across different experimental settings, making direct architecture comparisons approximate. Graph-based approaches are included only for coarse comparison (10 of 72 articles) and are not the focus of the analysis.
Reproducibility Details
Data
This is a systematic review, so no new models were trained. The authors collected metadata from 72 published articles. No datasets were generated or analyzed beyond the literature corpus.
Algorithms
Statistical comparisons used Mann-Whitney U tests for paired samples. Spearman correlation was used to assess the validity-novelty relationship. Outlier identification used the Valid/Sample (Validity x Novelty) metric with box plot analysis.
Evaluation
The review evaluates models using MOSES metrics: validity, uniqueness, novelty, scaffold diversity, scaffold novelty, fragment similarity, SNN, internal diversity, and FCD. Statistical tests were applied to compare medians across architecture families and between biased and unbiased models.
Hardware
Not applicable (systematic review, no model training performed).
Paper Information
Citation: Flores-Hernandez, H., & Martínez-Ledesma, E. (2024). A systematic review of deep learning chemical language models in recent era. Journal of Cheminformatics, 16(1), 129. https://doi.org/10.1186/s13321-024-00916-y
@article{floreshernandez2024systematic,
title={A systematic review of deep learning chemical language models in recent era},
author={Flores-Hernandez, Hector and Mart{\'i}nez-Ledesma, Emmanuel},
journal={Journal of Cheminformatics},
volume={16},
number={1},
pages={129},
year={2024},
publisher={BioMed Central},
doi={10.1186/s13321-024-00916-y}
}