A Foundational Systematization of Inverse Molecular Design
This paper is a Systematization of the nascent field of inverse molecular design using machine learning generative models. Published in Science in 2018, it organizes and contextualizes the rapidly emerging body of work on using deep generative models (variational autoencoders, generative adversarial networks, and reinforcement learning) to navigate chemical space and propose novel molecules with targeted properties. Rather than introducing a new method, the paper synthesizes the conceptual framework connecting molecular representations, generative architectures, and inverse design objectives, establishing a reference point for the field at a critical early stage.
The Challenge of Navigating Chemical Space
The core problem is the sheer scale of chemical space. For pharmacologically relevant small molecules alone, the number of possible structures is estimated at $10^{60}$. Traditional approaches to materials discovery rely on trial and error or high-throughput virtual screening (HTVS), both of which are fundamentally limited by the need to enumerate and evaluate candidates from a predefined library.
The conventional materials discovery pipeline, from concept to commercial product, historically takes 15 to 20 years, involving iterative cycles of simulation, synthesis, device integration, and characterization. Inverse design offers a conceptual alternative: start from a desired functionality and search for molecular structures that satisfy it. This inverts the standard paradigm where a molecule is proposed first and its properties are computed or measured afterward.
The key distinction the authors draw is between discriminative and generative models. A discriminative model learns $p(y|x)$, the conditional probability of properties $y$ given a molecule $x$. A generative model instead learns the joint distribution $p(x,y)$, which can be conditioned to yield either the direct design problem $p(y|x)$ or the inverse design problem $p(x|y)$.
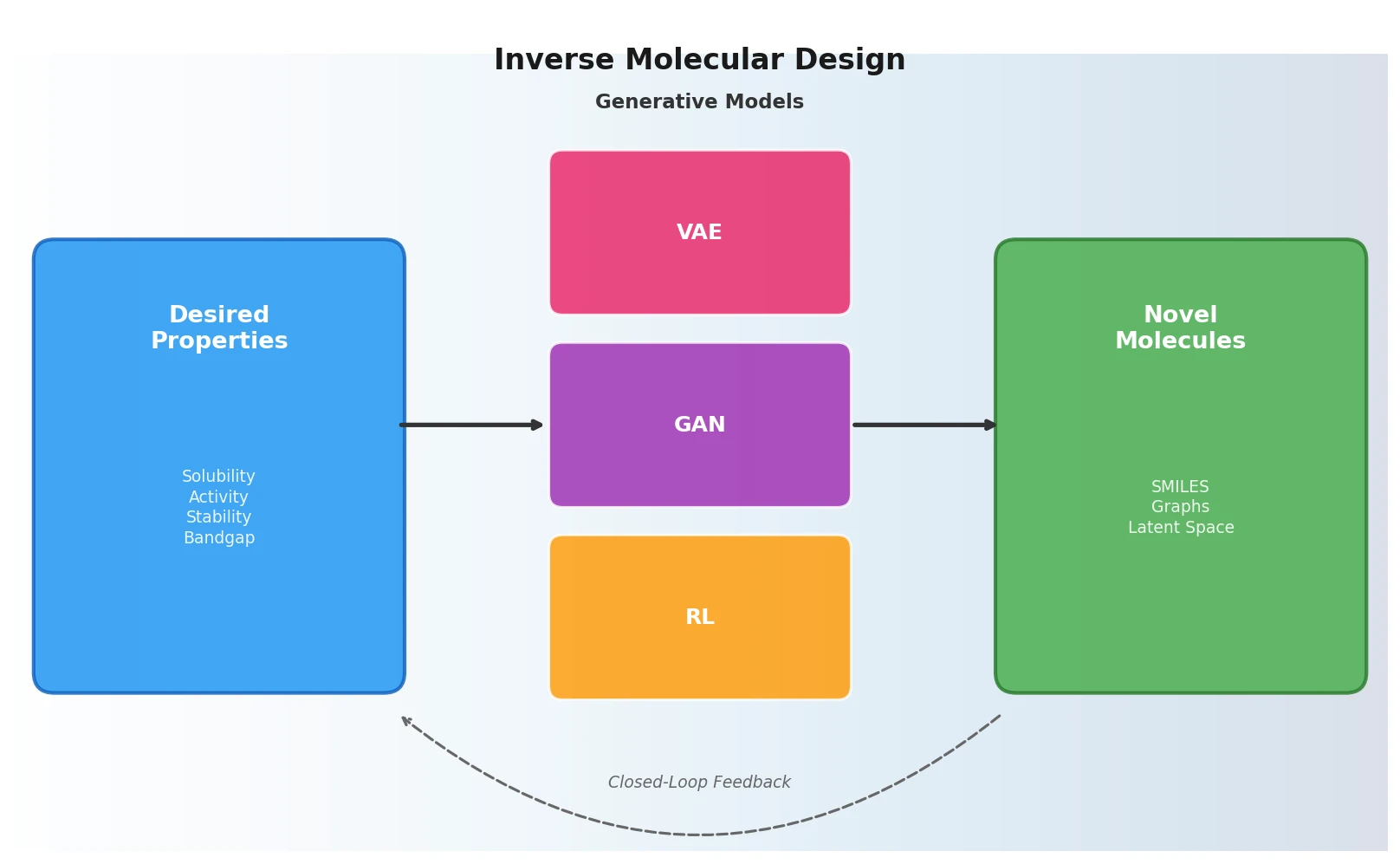
Three Pillars: VAEs, GANs, and Reinforcement Learning
The review organizes inverse molecular design approaches around three generative paradigms and the molecular representations they operate on.
Molecular Representations
The paper surveys representations across three broad categories:
- Discrete (text-based): SMILES strings encode molecular structure as 1D text following a grammar syntax. Their adoption has been driven by the availability of NLP deep learning tools.
- Continuous (vectors/tensors): Coulomb matrices, bag of bonds, fingerprints, symmetry functions, and electronic density representations. These expose different physical symmetries (permutational, rotational, reflectional, translational invariance).
- Weighted graphs: Molecules as undirected graphs where atoms are nodes and bonds are edges, with vectorized features on edges and nodes (bonding type, aromaticity, charge, distance).
An ideal representation for inverse design should be invertible, meaning it supports mapping back to a synthesizable molecular structure. SMILES strings and molecular graphs are invertible, while many continuous representations require lookup tables or auxiliary methods.
Variational Autoencoders (VAEs)
VAEs encode molecules into a continuous latent space and decode latent vectors back to molecular representations. The key insight is that by constraining the encoder to produce latent vectors following a Gaussian distribution, the model gains the ability to interpolate between molecules and sample novel structures. The latent space encodes a geometry: nearby points decode to similar molecules, and gradient-based optimization over this continuous space enables direct property optimization.
The VAE loss function combines a reconstruction term with a KL divergence regularizer:
$$\mathcal{L} = \mathbb{E}_{q(z|x)}[\log p(x|z)] - D_{KL}(q(z|x) | p(z))$$
where $q(z|x)$ is the encoder (approximate posterior), $p(x|z)$ is the decoder, and $p(z)$ is the prior (typically Gaussian).
Semi-supervised variants jointly train on molecules and properties, reorganizing latent space so molecules with similar properties cluster together. Gomez-Bombarelli et al. demonstrated local and global optimization across generated distributions using Bayesian optimization over latent space.
The review traces the evolution from character-level SMILES VAEs to grammar-aware and syntax-directed variants that improve the generation of syntactically valid structures.
Generative Adversarial Networks (GANs)
GANs pit a generator against a discriminator in an adversarial training framework. The generator learns to produce synthetic molecules from noise, while the discriminator learns to distinguish synthetic from real molecules. Training convergence for GANs is challenging, suffering from mode collapse and generator-discriminator imbalance.
For molecular applications, dealing with discrete SMILES data introduces nondifferentiability, addressed through workarounds like SeqGAN’s policy gradient approach and boundary-seeking GANs.
Reinforcement Learning (RL)
RL treats molecule generation as a sequential decision process where an agent (the generator) takes actions (adding characters to a SMILES string) to maximize a reward (desired molecular properties). Since rewards can only be assigned after sequence completion, Monte Carlo Tree Search (MCTS) is used to simulate possible completions and weight paths based on their success.
Applications include generation of drug-like molecules and retrosynthesis planning. Notable examples cited include RL for optimizing putative JAK2 inhibitors and molecules active against dopamine receptor type 2.
Hybrid Approaches
The review highlights that these paradigms are not exclusive. Examples include druGAN (adversarial autoencoder) and ORGANIC (combined GAN and RL), which leverage strengths of multiple frameworks.
Survey of Applications and Design Paradigms
Being a review paper, this work does not present new experiments but surveys existing applications across domains:
Drug Discovery: Most generative model applications at the time of writing targeted pharmaceutical properties, including solubility, melting temperature, synthesizability, and target activity. Popova et al. optimized for JAK2 inhibitors, and Olivecrona et al. targeted dopamine receptor type 2.
Materials Science: HTVS had been applied to organic photovoltaics (screening by frontier orbital energies and conversion efficiency), organic redox flow batteries (redox potential and solubility), organic LEDs (singlet-triplet gap), and inorganic materials via the Materials Project.
Chemical Space Exploration: Evolution strategies had been applied to map chemical space, with structured search procedures incorporating genotype representations and mutation operations. Bayesian sampling with sequential Monte Carlo and gradient-based optimization of properties with respect to molecular systems represented alternative inverse design strategies.
Graph-Based Generation: The paper notes the emerging extension of VAEs to molecular graphs (junction tree VAE) and message passing networks for incremental graph construction, though the graph isomorphism approximation problem remained a practical challenge.
Future Directions and Open Challenges
The authors identify several open directions for the field:
Closed-Loop Discovery: The ultimate goal is to concurrently propose, create, and characterize new materials with simultaneous data flow between components. At the time of writing, very few examples of successful closed-loop approaches existed.
Active Learning: Combining inverse design with Bayesian optimization enables models that adapt as they explore chemical space, expanding in regions of high uncertainty and discovering molecular regions with desirable properties as a function of composition.
Representation Learning: No single molecular representation works optimally for all properties. Graph and hierarchical representations were identified as areas needing further study. Representations that encode relevant physics tend to generalize better.
Improved Architectures: Memory-augmented sequence generation models, Riemannian optimization methods exploiting latent space geometry, multi-level VAEs for structured latent spaces, and inverse RL for learning reward functions were highlighted as promising research directions.
Integration into Education: The authors advocate for integrating ML into curricula across chemical, biochemical, medicinal, and materials sciences.
Limitations
As a review paper from 2018, this work captures the field at an early stage. Several limitations are worth noting:
- The survey is dominated by SMILES-based approaches, reflecting the state of the field at the time. Graph-based and 3D-aware generative models were just emerging.
- Quantitative benchmarking of generative models was not yet standardized. The review does not provide systematic comparisons across methods.
- The synthesis feasibility of generated molecules receives limited attention. The gap between computationally generated candidates and experimentally realizable molecules was (and remains) a significant challenge.
- Transformer-based architectures, which would come to dominate chemical language modeling, are not discussed, as the Transformer had only been published the year prior.
Reproducibility Details
As a review/perspective paper, this work does not introduce new models, datasets, or experiments. The reproducibility assessment applies to the cited primary works rather than the review itself.
Key Cited Methods and Their Resources
| Method | Authors | Type | Availability |
|---|---|---|---|
| Automatic Chemical Design (VAE) | Gomez-Bombarelli et al. | Code + Data | Published in ACS Central Science |
| Grammar VAE | Kusner et al. | Code | arXiv:1703.01925 |
| Junction Tree VAE | Jin et al. | Code | arXiv:1802.04364 |
| ORGANIC | Sanchez-Lengeling et al. | Code | ChemRxiv preprint |
| SeqGAN | Yu et al. | Code | AAAI 2017 |
| Neural Message Passing | Gilmer et al. | Code | arXiv:1704.01212 |
Paper Information
Citation: Sánchez-Lengeling, B., & Aspuru-Guzik, A. (2018). Inverse molecular design using machine learning: Generative models for matter engineering. Science, 361(6400), 360-365. https://doi.org/10.1126/science.aat2663
@article{sanchez-lengeling2018inverse,
title={Inverse molecular design using machine learning: Generative models for matter engineering},
author={S{\'a}nchez-Lengeling, Benjamin and Aspuru-Guzik, Al{\'a}n},
journal={Science},
volume={361},
number={6400},
pages={360--365},
year={2018},
publisher={American Association for the Advancement of Science},
doi={10.1126/science.aat2663}
}