A Systematization of Generative AI for Drug Design
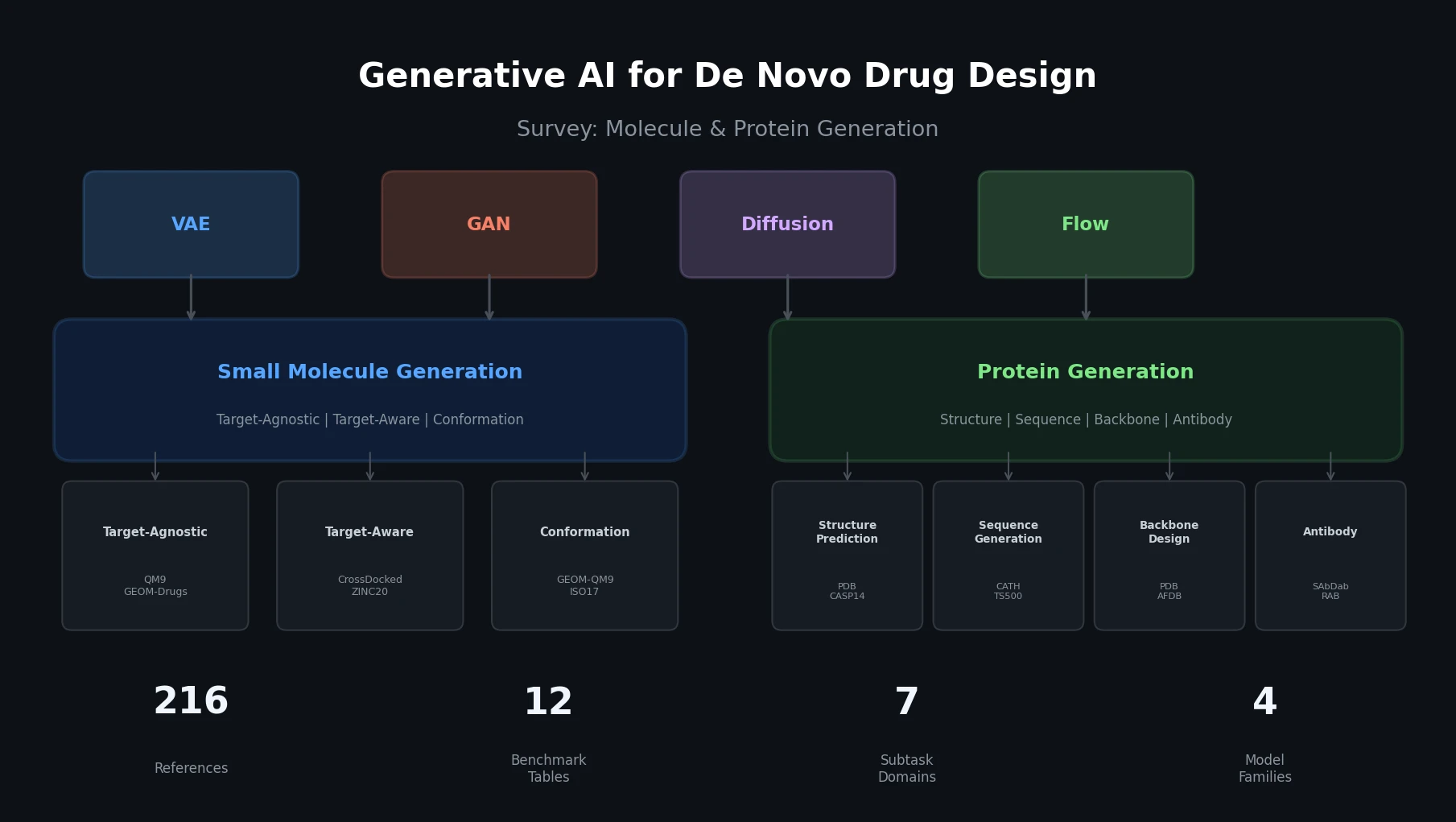
This is a Systematization paper that provides a broad survey of generative AI methods applied to de novo drug design. The survey organizes the field into two overarching themes: small molecule generation and protein generation. Within each theme, the authors identify subtasks, catalog datasets and benchmarks, describe model architectures, and compare the performance of leading methods using standardized metrics. The paper covers over 200 references and provides 12 comparative benchmark tables.
The primary contribution is a unified organizational framework that allows both micro-level comparisons within each subtask and macro-level observations across the two application domains. The authors highlight parallel developments in both fields, particularly the shift from sequence-based to structure-based approaches and the growing dominance of diffusion models.
The Challenge of Navigating De Novo Drug Design
The drug design process requires creating ligands that interact with specific biological targets. These range from small molecules (tens of atoms) to large proteins (monoclonal antibodies). Traditional discovery methods are computationally expensive, with preclinical trials costing hundreds of millions of dollars and taking 3-6 years. The chemical space of potential drug-like compounds is estimated at $10^{23}$ to $10^{60}$, making brute-force exploration infeasible.
AI-driven generative methods have gained traction in recent years, with over 150 AI-focused biotech companies initiating small-molecule drugs in the discovery phase and 15 in clinical trials. The rate of AI-fueled drug design processes has expanded by almost 40% each year.
The rapid development of the field, combined with its inherent complexity, creates barriers for new researchers. Several prior surveys exist, but they focus on specific aspects: molecule generation, protein generation, antibody generation, or specific model architectures like diffusion models. This survey takes a broader approach, covering both molecule and protein generation under a single organizational framework.
Unified Taxonomy: Two Themes, Seven Subtasks
The survey’s core organizational insight is structuring de novo drug design into two themes with distinct subtasks, while identifying common architectural patterns across them.
Generative Model Architectures
The survey covers four main generative model families used across both molecule and protein generation:
Variational Autoencoders (VAEs) encode inputs into a latent distribution and decode from sampled points. The encoder maps input $x$ to a distribution parameterized by mean $\mu_\phi(x)$ and variance $\sigma^2_\phi(x)$. Training minimizes reconstruction loss plus KL divergence:
$$\mathcal{L} = \mathcal{L}_{\text{recon}} + \beta \mathcal{L}_{\text{KL}}$$
where the KL loss is:
$$\mathcal{L}_{\text{KL}} = -\frac{1}{2} \sum_{k} \left(1 + \log(\sigma_k^{(i)2}) - \mu_k^{(i)2} - \sigma_k^{(i)2}\right)$$
Generative Adversarial Networks (GANs) use a generator-discriminator game. The generator $G$ creates instances from random noise $z$ sampled from a prior $p_z(z)$, while the discriminator $D$ distinguishes real from synthetic data:
$$\min_{G} \max_{D} \mathbb{E}_x[\log D(x; \theta_d)] + \mathbb{E}_{z \sim p(z)}[\log(1 - D(G(z; \theta_g); \theta_d))]$$
Flow-Based Models generate data by applying an invertible function $f: z_0 \mapsto x$ to transform a simple latent distribution (Gaussian) to the target distribution. The log-likelihood is computed using the change-of-variable formula:
$$\log p(x) = \log p_0(z) + \log \left| \det \frac{\partial f}{\partial z} \right|$$
Diffusion Models gradually add Gaussian noise over $T$ steps in a forward process and learn to reverse the noising via a denoising neural network. The forward step is:
$$x_{t+1} = \sqrt{1 - \beta_t} x_t + \sqrt{\beta_t} \epsilon, \quad \epsilon \sim \mathcal{N}(0, I)$$
The training loss minimizes the difference between the true noise and the predicted noise:
$$L_t = \mathbb{E}_{t \sim [1,T], x_0, \epsilon_t} \left[ | \epsilon_t - \epsilon_\theta(x_t, t) |^2 \right]$$
Graph neural networks (GNNs), particularly equivariant GNNs (EGNNs), are commonly paired with these generative methods to handle 2D/3D molecular and protein inputs. Diffusion and flow-based models are often paired with GNNs for processing 2D/3D-based input, while VAEs and GANs are typically used for 1D input.
Small Molecule Generation: Tasks, Datasets, and Models
Target-Agnostic Molecule Design
The goal is to generate a set of novel, valid, and stable molecules without conditioning on any specific biological target. Models are evaluated on atom stability, molecule stability, validity, uniqueness, novelty, and QED (Quantitative Estimate of Drug-Likeness).
Datasets: QM9 (small stable molecules from GDB-17) and GEOM-Drug (more complex, drug-like molecules).
The field has shifted from SMILES-based VAEs (CVAE, GVAE, SD-VAE) to 2D graph methods (JTVAE) and then to 3D diffusion-based models. Current leading methods on QM9:
| Model | Type | At Stb. (%) | Mol Stb. (%) | Valid (%) | Val/Uniq. (%) |
|---|---|---|---|---|---|
| MiDi | EGNN, Diffusion | 99.8 | 97.5 | 97.9 | 97.6 |
| MDM | EGNN, VAE, Diffusion | 99.2 | 89.6 | 98.6 | 94.6 |
| JODO | EGNN, Diffusion | 99.2 | 93.4 | 99.0 | 96.0 |
| GeoLDM | VAE, Diffusion | 98.9 | 89.4 | 93.8 | 92.7 |
| EDM | EGNN, Diffusion | 98.7 | 82.0 | 91.9 | 90.7 |
EDM provided an initial baseline using diffusion with an equivariant GNN. GCDM introduced attention-based geometric message-passing. MDM separately handles covalent bond edges and Van der Waals forces, and also addresses diversity through an additional distribution-controlling noise variable. GeoLDM maps molecules to a lower-dimensional latent space for more efficient diffusion. MiDi uses a “relaxed” EGNN and jointly models 2D and 3D information through a graph representation capturing both spatial and connectivity data.
On the larger GEOM-Drugs dataset, performance drops for most models:
| Model | At Stb. (%) | Mol Stb. (%) | Valid (%) | Val/Uniq. (%) |
|---|---|---|---|---|
| MiDi | 99.8 | 91.6 | 77.8 | 77.8 |
| MDM | – | 62.2 | 99.5 | 99.0 |
| GeoLDM | 84.4 | – | 99.3 | – |
| EDM | 81.3 | – | – | – |
MiDi distinguishes itself for generating more stable complex molecules, though at the expense of validity. Models generally perform well on QM9 but show room for improvement on more complex GEOM-Drugs molecules.
Target-Aware Molecule Design
Target-aware generation produces molecules for specific protein targets, using either ligand-based (LBDD) or structure-based (SBDD) approaches. SBDD methods have become more prevalent as protein structure information becomes increasingly available.
Datasets: CrossDocked2020 (22.5M ligand-protein pairs), ZINC20, Binding MOAD.
Metrics: Vina Score (docking energy), High Affinity Percentage, QED, SA Score (synthetic accessibility), Diversity (Tanimoto similarity).
| Model | Type | Vina | Affinity (%) | QED | SA | Diversity |
|---|---|---|---|---|---|---|
| DiffSBDD | EGNN, Diffusion | -7.333 | – | 0.467 | 0.554 | 0.758 |
| Luo et al. | SchNet | -6.344 | 29.09 | 0.525 | 0.657 | 0.720 |
| TargetDiff | EGNN, Diffusion | -6.3 | 58.1 | 0.48 | 0.58 | 0.72 |
| LiGAN | CNN, VAE | -6.144 | 21.1 | 0.39 | 0.59 | 0.66 |
| Pocket2Mol | EGNN, MLP | -5.14 | 48.4 | 0.56 | 0.74 | 0.69 |
DrugGPT is an LBDD autoregressive model using transformers on tokenized protein-ligand pairs. Among the SBDD models, LiGAN introduces a 3D CNN-VAE framework, Pocket2Mol emphasizes binding pocket geometry using an EGNN with geometric vector MLP layers, and Luo et al. model atomic probabilities in the binding site using SchNet. TargetDiff performs diffusion on an EGNN and optimizes binding affinity by reflecting low atom type entropy. DiffSBDD applies an inpainting approach by masking and replacing segments of ligand-protein complexes. DiffSBDD leads in Vina score and diversity, while TargetDiff leads in high affinity. Interestingly, diffusion-based methods are outperformed by Pocket2Mol on drug-likeness metrics (QED and SA).
Molecular Conformation Generation
Conformation generation involves producing 3D structures from 2D connectivity graphs. Models are evaluated on Coverage (COV, percentage of ground-truth conformations “covered” within an RMSD threshold) and Matching (MAT, average RMSD to closest ground-truth conformation).
Datasets: GEOM-QM9, GEOM-Drugs, ISO17.
| Model | Type | GEOM-QM9 COV (%) | GEOM-QM9 MAT | GEOM-Drugs COV (%) | GEOM-Drugs MAT |
|---|---|---|---|---|---|
| Torsional Diff. | Diffusion | 92.8 | 0.178 | 72.7* | 0.582 |
| DGSM | MPNN, Diffusion | 91.49 | 0.2139 | 78.73 | 1.0154 |
| GeoDiff | GFN, Diffusion | 90.07 | 0.209 | 89.13 | 0.8629 |
| ConfGF | GIN, Diffusion | 88.49 | 0.2673 | 62.15 | 1.1629 |
| GeoMol | MPNN | 71.26 | 0.3731 | 67.16 | 1.0875 |
*Torsional Diffusion uses a 0.75 A threshold instead of the standard 1.25 A for GEOM-Drugs coverage, leading to a deflated score. It outperforms GeoDiff and GeoMol when evaluated at the same threshold.
Torsional Diffusion operates in the space of torsion angles rather than Cartesian coordinates, allowing for improved representation and fewer denoising steps. GeoDiff uses Euclidean-space diffusion, treating each atom as a particle and incorporating Markov kernels that preserve E(3) equivariance through a graph field network (GFN) layer.
Protein Generation: From Sequence to Structure
Protein Representation Learning
Representation learning creates embeddings for protein inputs to support downstream tasks. Models are evaluated on contact prediction, fold classification (at family, superfamily, and fold levels), and stability prediction (Spearman’s $\rho$).
Key models include: UniRep (mLSTM RNN), ProtBERT (BERT applied to amino acid sequences), ESM-1B (33-layer, 650M parameter transformer), MSA Transformer (pre-trained on MSA input), and GearNET (Geo-EGNN using 3D structure with directed edges). OntoProtein and KeAP incorporate knowledge graphs for direct knowledge injection.
Protein Structure Prediction
Given an amino acid sequence, models predict 3D point coordinates for each residue. Evaluated using RMSD, GDT-TS, TM-score, and LDDT on CASP14 and CAMEO benchmarks.
AlphaFold2 is the landmark model, integrating MSA and pair representations through transformers with invariant point attention (IPA). ESMFold uses ESM-2 language model representations instead of MSAs, achieving faster processing. RoseTTAFold uses a three-track neural network learning from 1D sequence, 2D distance map, and 3D backbone coordinate information simultaneously. EigenFold uses diffusion, representing the protein as a system of harmonic oscillators.
| Model | Type | CAMEO RMSD | CAMEO TMScore | CAMEO GDT-TS | CAMEO lDDT | CASP14 TMScore |
|---|---|---|---|---|---|---|
| AlphaFold2 | Transformer | 3.30 | 0.87 | 0.86 | 0.90 | 0.38 |
| ESMFold | Transformer | 3.99 | 0.85 | 0.83 | 0.87 | 0.68 |
| RoseTTAFold | Transformer | 5.72 | 0.77 | 0.71 | 0.79 | 0.37 |
| EigenFold | Diffusion | 7.37 | 0.75 | 0.71 | 0.78 | – |
Sequence Generation (Inverse Folding)
Given a fixed protein backbone structure, models generate amino acid sequences that will fold into that structure. The space of valid sequences is between $10^{65}$ and $10^{130}$.
Evaluated using Amino Acid Recovery (AAR), diversity, RMSD, nonpolar loss, and perplexity (PPL):
$$\text{PPL} = \exp\left(\frac{1}{N} \sum_{i=1}^{N} \log P(x_i | x_1, x_2, \ldots x_{i-1})\right)$$
ProteinMPNN is the current top performer, generating the most accurate sequences and leading in AAR, RMSD, and nonpolar loss. It uses a message-passing neural network with a flexible, order-agnostic autoregressive approach.
| Model | Type | AAR (%) | Div. | RMSD | Non. | Time (s) |
|---|---|---|---|---|---|---|
| ProteinMPNN | MPNN | 48.7 | 0.168 | 1.019 | 1.061 | 112 |
| ESM-IF1 | Transformer | 47.7 | 0.184 | 1.265 | 1.201 | 1980 |
| GPD | Transformer | 46.2 | 0.219 | 1.758 | 1.333 | 35 |
| ABACUS-R | Transformer | 45.7 | 0.124 | 1.482 | 0.968 | 233280 |
| 3D CNN | CNN | 44.5 | 0.272 | 1.62 | 1.027 | 536544 |
| PiFold | GNN | 42.8 | 0.141 | 1.592 | 1.464 | 221 |
| ProteinSolver | GNN | 24.6 | 0.186 | 5.354 | 1.389 | 180 |
Results are from the independent benchmark by Yu et al. GPD remains the fastest method, generating sequences around three times faster than ProteinMPNN. Current SOTA models recover fewer than half of target amino acid residues, indicating room for improvement.
Backbone Design
Backbone design creates protein structures from scratch, representing the core of de novo protein design. Models generate coordinates for backbone atoms (nitrogen, alpha-carbon, carbonyl, oxygen) and use external tools like Rosetta for side-chain packing.
Two evaluation paradigms exist: context-free generation (evaluated by self-consistency TM, or scTM) and context-given generation (inpainting, evaluated by AAR, PPL, RMSD).
ProtDiff represents residues as 3D Cartesian coordinates and uses particle-filtering diffusion. FoldingDiff instead uses an angular representation (six angles per residue) with a BERT-based DDPM. LatentDiff embeds proteins into a latent space using an equivariant autoencoder, then applies equivariant diffusion, analogous to GeoLDM for molecules. These early models work well for short proteins (up to 128 residues) but struggle with longer structures.
Frame-based methods address this scaling limitation. Genie uses Frenet-Serret frames with paired residue representations and IPA for noise prediction. FrameDiff parameterizes backbone structures on the $SE(3)^N$ manifold of frames using a score-based generative model. RFDiffusion is the current leading model, combining RoseTTAFold structure prediction with diffusion. It fine-tunes RoseTTAFold weights on a masked input sequence and random noise coordinates, using “self-conditioning” on predicted structures. Protpardelle co-designs sequence and structure by creating a “superposition” over possible sidechain states and collapsing them during each iterative diffusion step.
| Model | Type | scTM (%) | Design. (%) | PPL | AAR (%) | RMSD |
|---|---|---|---|---|---|---|
| RFDiffusion | Diffusion | – | 95.1 | – | – | – |
| Protpardelle | Diffusion | 85 | – | – | – | – |
| FrameDiff | Diffusion | 84 | 48.3 | – | – | – |
| Genie | Diffusion | 81.5 | 79.0 | – | – | – |
| LatentDiff | EGNN, Diffusion | 31.6 | – | – | – | – |
| FoldingDiff | Diffusion | 14.2 | – | – | – | – |
| ProtDiff | EGNN, Diffusion | 11.8 | – | – | 12.47* | 8.01* |
*ProtDiff context-given results are tested only on beta-lactamase metalloproteins from PDB.
Antibody Design
The survey covers antibody structure prediction, representation learning, and CDR-H3 generation. Antibodies are Y-shaped proteins with complementarity-determining regions (CDRs), where CDR-H3 is the most variable and functionally important region.
For CDR-H3 generation, models have progressed from sequence-based (LSTM) to structure-based (RefineGNN) and sequence-structure co-design approaches (MEAN, AntiDesigner, DiffAb). dyMEAN is the current leading model, providing an end-to-end method incorporating structure prediction, docking, and CDR generation into a single framework. MSA alignment cannot be used for antibody input, which makes general models like AlphaFold2 inefficient for antibody prediction. Specialized models like IgFold use sequence embeddings from AntiBERTy with invariant point attention to achieve faster antibody structure prediction.
Peptide Design
The survey briefly covers peptide generation, including models for therapeutic peptide generation (MMCD), peptide-protein interaction prediction (PepGB), peptide representation learning (PepHarmony), peptide sequencing (AdaNovo), and signal peptide prediction (PEFT-SP).
Current Trends, Challenges, and Future Directions
Current Trends
The survey identifies several parallel trends across molecule and protein generation:
Shift from sequence to structure: In molecule generation, graph-based diffusion models (GeoLDM, MiDi, TargetDiff) now dominate. In protein generation, structure-based representation learning (GearNET) and diffusion-based backbone design (RFDiffusion) have overtaken sequence-only methods.
Dominance of E(3) equivariant architectures: EGNNs appear across nearly all subtasks, reflecting the physical requirement that molecular and protein properties should be invariant to rotation and translation.
Structure-based over ligand-based approaches: In target-aware molecule design, SBDD methods that use 3D protein structures demonstrate clear advantages over LBDD approaches that operate on amino acid sequences alone.
Challenges
For small molecule generation:
- Complexity: Models perform well on simple QM9 but struggle with complex GEOM-Drugs molecules.
- Applicability: Generating molecules with high binding affinity to targets remains difficult.
- Explainability: Methods are black-box, offering no insight into why generated molecules have desired properties.
For protein generation:
- Benchmarking: Protein generative tasks lack a standard evaluative procedure, with variance between each model’s metrics and testing conditions.
- Performance: SOTA models still struggle with fold classification, gene ontology, and antibody CDR-H3 generation.
The authors also note that many generative tasks are evaluated using predictive models (e.g., classifier networks for binding affinity or molecular properties). Improvements to these classification methods would lead to more precise alignment with real-world biological applications.
Future Directions
The authors identify increasing performance in existing tasks, defining more applicable tasks (especially in molecule-protein binding, antibody generation), and exploring entirely new areas of research as key future directions.
Reproducibility Details
As a survey paper, this work does not produce new models, datasets, or experimental results. All benchmark numbers reported are from the original papers cited.
Data
The survey catalogs the following key datasets across subtasks:
| Subtask | Datasets | Notes |
|---|---|---|
| Target-agnostic molecule | QM9, GEOM-Drug | QM9 from GDB-17; GEOM-Drug for complex molecules |
| Target-aware molecule | CrossDocked2020, ZINC20, Binding MOAD | CrossDocked2020 most used (22.5M pairs) |
| Conformation generation | GEOM-QM9, GEOM-Drugs, ISO17 | Conformer sets for molecules |
| Protein structure prediction | PDB, CASP14, CAMEO | CASP biennial blind evaluation |
| Protein sequence generation | PDB, UniRef, UniParc, CATH, TS500 | CATH for domain classification |
| Backbone design | PDB, AlphaFoldDB, SCOP, CATH | AlphaFoldDB for expanded structural coverage |
| Antibody structure | SAbDab, RAB | SAbDab: all antibody structures from PDB |
| Antibody CDR generation | SAbDab, RAB, SKEMPI | SKEMPI for affinity optimization |
Artifacts
| Artifact | Type | License | Notes |
|---|---|---|---|
| GenAI4Drug | Code | Not specified | Organized repository of all covered sources |
Paper Information
Citation: Tang, X., Dai, H., Knight, E., Wu, F., Li, Y., Li, T., & Gerstein, M. (2024). A survey of generative AI for de novo drug design: New frontiers in molecule and protein generation. Briefings in Bioinformatics, 25(4), bbae338. https://doi.org/10.1093/bib/bbae338
Publication: Briefings in Bioinformatics, Volume 25, Issue 4, 2024.
Additional Resources:
@article{tang2024survey,
title={A survey of generative AI for de novo drug design: new frontiers in molecule and protein generation},
author={Tang, Xiangru and Dai, Howard and Knight, Elizabeth and Wu, Fang and Li, Yunyang and Li, Tianxiao and Gerstein, Mark},
journal={Briefings in Bioinformatics},
volume={25},
number={4},
pages={bbae338},
year={2024},
doi={10.1093/bib/bbae338}
}