A Systematization of Foundation Models for Chemistry
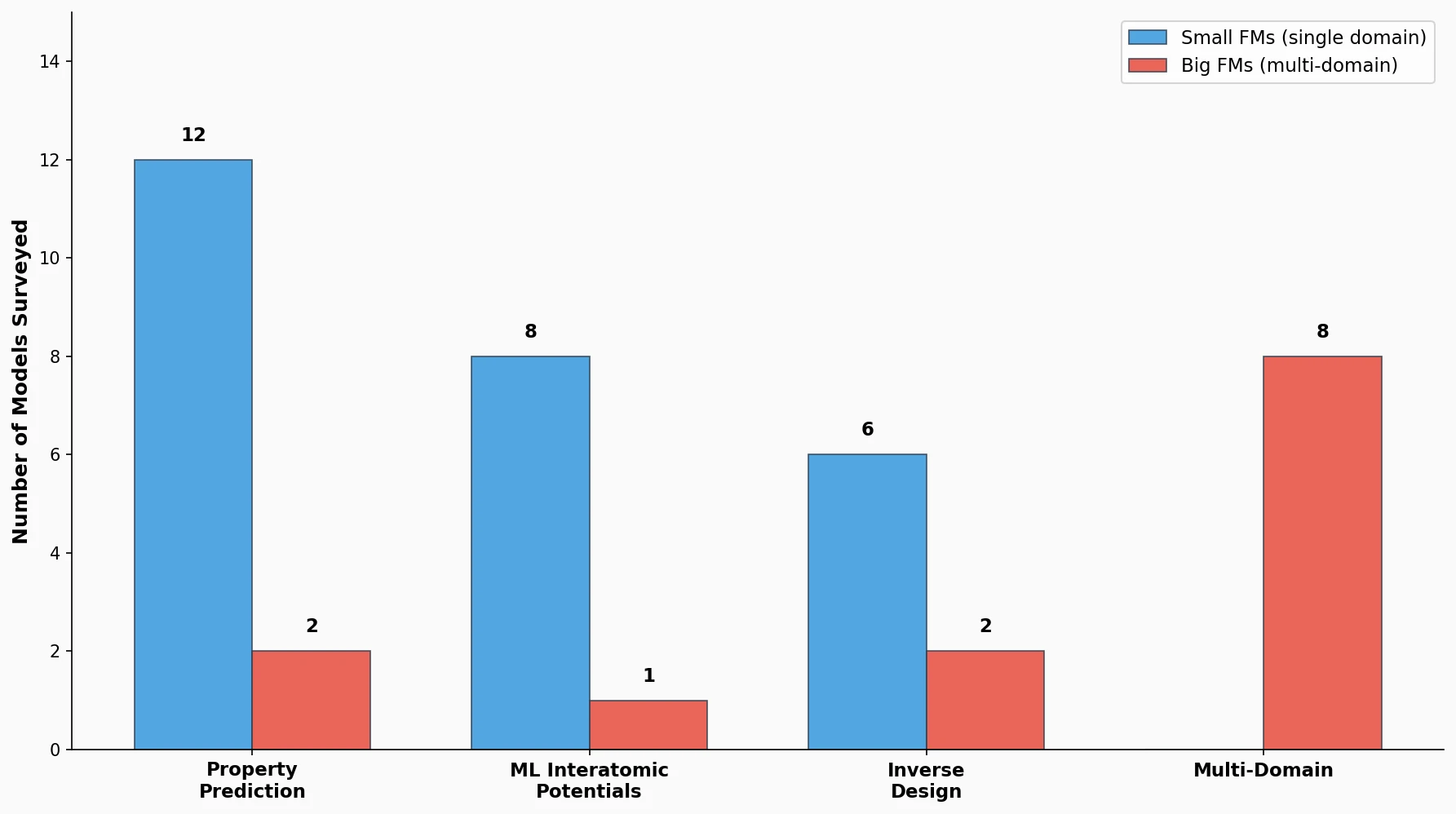
This is a Systematization paper. It organizes the rapidly growing landscape of foundation models in chemistry into a coherent taxonomy. The paper distinguishes between “small” foundation models (pretrained for a single application domain) and “big” foundation models (adaptable across multiple domains such as property prediction and inverse design). It covers models based on graph neural networks (GNNs) and language models, reviews pretraining strategies (self-supervised, multimodal, supervised), and maps approximately 40 models across four application domains.
Why a Foundation Model Perspective for Chemistry?
Foundation models have transformed NLP and computer vision through large-scale pretraining and transfer learning. In chemistry, however, several persistent challenges motivate the adoption of this paradigm:
- Data scarcity: Chemical datasets are often small and expensive to generate (requiring experiments or quantum mechanical calculations), unlike the large annotated datasets available in NLP/CV.
- Poor generalization: ML models in chemistry frequently need to extrapolate to out-of-domain compounds (e.g., novel drug candidates, unseen crystal structures), where conventional models struggle.
- Limited transferability: Traditional ML interatomic potentials (MLIPs) are trained on system-specific datasets and cannot be easily transferred across different chemical systems.
Foundation models address these by learning general representations from large unlabeled datasets, which can then be adapted to specific downstream tasks via finetuning. The paper argues that summarizing this fast-moving field is timely, given the diversity of approaches emerging across molecular property prediction, MLIPs, inverse design, and multi-domain applications.
Small vs. Big Foundation Models: A Two-Tier Taxonomy
The paper’s central organizing framework distinguishes two scopes of foundation model:
Small foundation models are pretrained models adapted to various tasks within a single application domain. Examples include:
- A model pretrained on large molecular databases that predicts multiple molecular properties (band gap, formation energy, etc.)
- A universal MLIP that can simulate diverse chemical systems
- A pretrained generative model adapted for inverse design of different target properties
Big foundation models span multiple application domains, handling both property prediction and inverse design within a single framework. These typically use multimodal learning (combining SMILES/graphs with text) or build on large language models.
Architectures
The paper reviews two primary architecture families:
Graph Neural Networks (GNNs) represent molecules and crystals as graphs $G = (V, E)$ with nodes (atoms) and edges (bonds). Node features are updated through message passing:
$$ m_{i}^{t+1} = \sum_{j \in N(i)} M_{t}(v_{i}^{t}, v_{j}^{t}, e_{ij}^{t}) $$
$$ v_{i}^{t+1} = U_{t}(v_{i}^{t}, m_{i}^{t+1}) $$
After $T$ message-passing steps, a readout function produces a graph-level feature:
$$ g = R({v_{i}^{T} \mid i \in G}) $$
Recent equivariant GNNs (e.g., NequIP, MACE, EquformerV2) use vectorial features that respect geometric symmetries, improving expressivity for tasks sensitive to 3D structure.
Language Models operate on string representations of molecules (SMILES, SELFIES) or crystal structures. Autoregressive models like GPT maximize:
$$ \prod_{t=1}^{T} P(y_{t} \mid x_{1}, x_{2}, \ldots, x_{t-1}) $$
Transformers use self-attention:
$$ \text{Attention}(Q, K, V) = \text{softmax}\left(\frac{QK^{T}}{\sqrt{d_{k}}}\right)V $$
Pretraining Strategies
The paper categorizes pretraining methods into three self-supervised learning (SSL) approaches plus supervised and multimodal strategies:
| Strategy | Mechanism | Example Models |
|---|---|---|
| Contrastive learning | Maximize similarity between positive pairs, minimize for negatives | GraphCL, MolCLR, GraphMVP, CrysGNN |
| Predictive learning | Predict self-generated labels (node context, functional groups, space group) | GROVER, Hu et al., CrysGNN |
| Generative learning | Reconstruct masked nodes/edges or entire molecules/SMILES | SMILES-BERT, ChemBERTa-2, MoLFormer |
| Supervised pretraining | Train on energy, forces, stress from DFT databases | M3GNet, CHGNet, MACE-MP-0, MatterSim |
| Multimodal learning | Learn joint representations across SMILES/graph + text modalities | KV-PLM, MoMu, MoleculeSTM, SPMM |
A common finding across studies is that combining local and global information (e.g., via contrastive learning between node-level and graph-level views, or supervised learning on both forces and total energy) produces more transferable representations.
Survey of Models Across Four Domains
Property Prediction
The paper reviews 13 models for molecular and materials property prediction. Key findings:
- Contrastive learning approaches (GraphCL, MolCLR, GraphMVP) achieve strong results by defining positive pairs through augmentation, 2D/3D structure views, or crystal system membership.
- Language model approaches (SMILES-BERT, ChemBERTa-2, MoLFormer) show that transformers trained on SMILES via masked language modeling can compete with GNN-based approaches.
- MoLFormer, pretrained on 1.1 billion SMILES from PubChem and ZINC, outperformed many baselines including GNNs on MoleculeNet and QM9 benchmarks. Its attention maps captured molecular structural features directly from SMILES strings.
- For crystalline materials, CrysGNN combined contrastive, predictive, and generative learning, demonstrating improvements even on small experimental datasets.
Machine Learning Interatomic Potentials (MLIPs)
The paper surveys 10 universal MLIPs, all using supervised learning on DFT-calculated energies, forces, and stresses:
| Model | Architecture | Training Data Size | Key Capability |
|---|---|---|---|
| M3GNet | GNN | 187K (MP) | First universal MLIP |
| CHGNet | GNN | 1.58M (MPtrj) | Predicts magnetic moments |
| MACE-MP-0 | MACE | 1.58M (MPtrj) | 35 diverse applications |
| GNoME potential | NequIP | 89M | Zero-shot comparable to trained MLIPs |
| MatterSim | M3GNet/Graphormer | 17M | SOTA on Matbench Discovery |
| eqV2 | EquformerV2 | 118M (OMat24) | Structural relaxation |
The GNoME potential, trained on approximately 89 million data points, achieved zero-shot performance comparable to state-of-the-art MLIPs trained from scratch. MatterSim, trained on over 17 million entries across wide temperature (0-5000K) and pressure (0-1000 GPa) ranges, achieved state-of-the-art on Matbench Discovery and accurately computed thermodynamic and lattice dynamic properties.
Inverse Design
Few pretrained generative models for inverse design exist. The paper highlights three:
- MatterGen (Microsoft): Diffusion model pretrained on Alexandria/MP databases (607K structures), finetuned for conditional generation on band gap, elastic modulus, spacegroup, and composition. Generated S.U.N. (stable, unique, novel) materials at rates more than 2x the previous state of the art.
- GP-MoLFormer (IBM): MoLFormer pretrained on 1.1B SMILES, finetuned via pair-tuning for property-guided molecular optimization.
- CrystalLLM: Finetuned LLaMA-2 70B for crystal generation with target spacegroup and composition using string representations and prompting.
Multi-Domain Models
The paper covers two multi-domain categories:
Property prediction + MLIP: Denoising pretraining learns virtual forces that guide noisy configurations back to equilibrium, connecting to force prediction. Joint multi-domain pretraining (JMP) from Meta FAIR achieved state-of-the-art on 34 of 40 tasks spanning molecules, crystals, and MOFs by training simultaneously on diverse energy/force databases.
Property prediction + inverse design: Multimodal models (KV-PLM, MoMu, MoleculeSTM, MolFM, SPMM) learn joint representations from molecular structures and text, enabling text-based inverse design and property prediction in a single framework. LLM-based models (ChemDFM, nach0, finetuned GPT-3) can interact with humans and handle diverse chemistry tasks through instruction tuning.
Trends and Future Directions
Scope Expansion
The authors identify three axes for expanding foundation model scope:
- Material types: Most models target molecules or a single material class. Foundation models that span molecules, crystals, surfaces, and MOFs could exploit shared chemistry across materials.
- Modalities: Beyond SMILES, graphs, and text, additional modalities (images, spectral data like XRD patterns) remain underexplored.
- Downstream tasks: Extending to new chemistry and tasks through emergent capabilities, analogous to the capabilities observed in LLMs at scale.
Performance and Scaling
Key scaling challenges include:
- Data quality vs. quantity: Noisy DFT labels (e.g., HOMO-LUMO gaps with high uncertainty from different functionals/basis sets) can limit scalability and out-of-distribution performance.
- GNN scalability: While transformers scale to hundreds of billions of parameters, GNNs have rarely been explored above one million parameters due to oversmoothing and the curse of dimensionality. Recent work by Sypetkowski et al. demonstrated scaling GNNs to 3 billion parameters with consistent improvements.
- Database integration: Combining datasets from different DFT codes requires proper alignment (e.g., total energy alignment methods).
Efficiency
For MLIPs, efficiency is critical since MD simulations require millions of inference steps. Approaches include:
- Knowledge distillation from expensive teacher models to lighter student models
- Model compression techniques (quantization, pruning) adapted for GNNs
- Investigating whether strict equivariance is always necessary
Interpretability
Foundation models can generate hallucinations or mode-collapsed outputs. The authors highlight recent interpretability advances (feature extraction from Claude 3, knowledge localization and editing in transformers) as promising directions for more reliable chemical applications.
Key Findings and Limitations
Key findings:
- Combining local and global information in pretraining consistently improves downstream performance across all domains reviewed.
- Self-supervised pretraining enables effective transfer learning even in low-data regimes, a critical advantage for chemistry.
- Universal MLIPs have reached the point where zero-shot performance can be comparable to system-specific trained models.
- Multimodal learning is the most promising approach for big foundation models capable of spanning property prediction and inverse design.
Limitations acknowledged by the authors:
- The precise definition of “foundation model” in chemistry is not established and varies by scope.
- Most surveyed models focus on molecules, with crystalline materials less explored.
- Benchmarks for low-data regimes and out-of-distribution performance are insufficient.
- The paper focuses on three domains (property prediction, MLIPs, inverse design) and does not cover retrosynthesis, reaction prediction, or other chemical tasks in depth.
Reproducibility Details
Data
This is a perspective/review paper. No new data or models are introduced. The paper surveys existing models and their training datasets, summarized in Table 1 of the paper.
Algorithms
Not applicable (review paper). The paper describes pretraining strategies (contrastive, predictive, generative, supervised, multimodal) at a conceptual level with references to the original works.
Models
Not applicable (review paper). The paper catalogs approximately 40 foundation models across four domains. See Table 1 in the paper for the complete listing.
Evaluation
Not applicable (review paper). The paper references benchmark results from the original studies (MoleculeNet, QM9, Matbench, Matbench Discovery, JARVIS-DFT) but does not perform independent evaluation.
Hardware
Not applicable (review paper).
Paper Information
Citation: Choi, J., Nam, G., Choi, J., & Jung, Y. (2025). A Perspective on Foundation Models in Chemistry. JACS Au, 5(4), 1499-1518. https://doi.org/10.1021/jacsau.4c01160
@article{choi2025perspective,
title={A Perspective on Foundation Models in Chemistry},
author={Choi, Junyoung and Nam, Gunwook and Choi, Jaesik and Jung, Yousung},
journal={JACS Au},
volume={5},
number={4},
pages={1499--1518},
year={2025},
publisher={American Chemical Society},
doi={10.1021/jacsau.4c01160}
}