A Systematization of Deep Generative Models for Molecular Design
This is a Systematization paper that organizes and compares the rapidly growing literature on deep generative modeling for molecules. Published in 2019, it catalogs 45 papers from the preceding two years, classifying them by architecture (RNNs, VAEs, GANs, reinforcement learning) and molecular representation (SMILES strings, context-free grammars, graph tensors, 3D voxels). The review provides mathematical foundations for each technique, identifies cross-cutting themes, and proposes a framework for reward function design that addresses diversity, novelty, stability, and synthesizability.
The Challenge of Navigating Vast Chemical Space
The space of potential drug-like molecules has been estimated to contain between $10^{23}$ and $10^{60}$ compounds, while only about $10^{8}$ have ever been synthesized. Traditional approaches to molecular design rely on combinatorial methods, mixing known scaffolds and functional groups, but these generate many unstable or unsynthesizable candidates. High-throughput screening (HTS) and virtual screening (HTVS) help but remain computationally expensive. The average cost to bring a new drug to market exceeds one billion USD, with a 13-year average timeline from discovery to market.
By 2016, deep generative models had shown strong results in producing original images, music, and text. The “molecular autoencoder” of Gomez-Bombarelli et al. (2016/2018) first applied these techniques to molecular generation, triggering an explosion of follow-up work. By the time of this review, the landscape had grown complex enough, with many architectures, representation schemes, and no agreed-upon benchmarking standards, to warrant systematic organization.
Molecular Representations and Architecture Taxonomy
The review’s core organizational contribution is a two-axis taxonomy: molecular representations on one axis and deep learning architectures on the other.
Molecular Representations
The review categorizes representations into 3D and 2D graph-based schemes:
3D representations include raw voxels (placing nuclear charges on a grid), smoothed voxels (Gaussian blurring around nuclei), and tensor field networks. These capture full geometric information but suffer from high dimensionality, sparsity, and difficulty encoding rotation/translation invariance.
2D graph representations include:
- SMILES strings: The dominant representation, encoding molecular graphs as ASCII character sequences via depth-first traversal. Non-unique (each molecule with $N$ heavy atoms has at least $N$ SMILES representations), but invertible and widely supported.
- Canonical SMILES: Unique but potentially encode grammar rules rather than chemical structure.
- Context-free grammars (CFGs): Decompose SMILES into grammar rules to improve validity rates, though not to 100%.
- Tensor representations: Store atom types in a vertex feature matrix $X \in \mathbb{R}^{N \times |\mathcal{A}|}$ and bond types in an adjacency tensor $A \in \mathbb{R}^{N \times N \times Y}$.
- Graph operations: Directly build molecular graphs by adding atoms and bonds, guaranteeing 100% chemical validity.
Deep Learning Architectures
Recurrent Neural Networks (RNNs) generate SMILES strings character by character, typically using LSTM or GRU units. Training uses maximum likelihood estimation (MLE) with teacher forcing:
$$ L^{\text{MLE}} = -\sum_{s \in \mathcal{X}} \sum_{t=2}^{T} \log \pi_{\theta}(s_{t} \mid S_{1:t-1}) $$
Thermal rescaling of the output distribution controls the diversity-validity tradeoff via a temperature parameter $T$. RNNs achieved SMILES validity rates of 94-98%.
Variational Autoencoders (VAEs) learn a continuous latent space by maximizing the evidence lower bound (ELBO):
$$ \mathcal{L}_{\theta,\phi}(x) = \mathbb{E}_{z \sim q_{\phi}(z|x)}[\log p_{\theta}(x|z)] - D_{\text{KL}}[q_{\phi}(z|x), p(z)] $$
The first term encourages accurate reconstruction while the KL divergence term regularizes the latent distribution toward a standard Gaussian prior $p(z) = \mathcal{N}(z, 0, I)$. Variants include grammar VAEs (GVAEs), syntax-directed VAEs, junction tree VAEs, and adversarial autoencoders (AAEs) that replace the KL term with adversarial training.
Generative Adversarial Networks (GANs) train a generator against a discriminator using the minimax objective:
$$ \min_{G} \max_{D} V(D, G) = \mathbb{E}_{x \sim p_{d}(x)}[\log D(x)] + \mathbb{E}_{z \sim p_{z}(z)}[\log(1 - D(G(z)))] $$
The review shows that with an optimal discriminator, the generator objective reduces to minimizing the Jensen-Shannon divergence, which captures both forward and reverse KL divergence terms. This provides a more “balanced” training signal than MLE alone. The Wasserstein GAN (WGAN) uses the Earth mover’s distance for more stable training:
$$ W(p, q) = \inf_{\gamma \in \Pi(p,q)} \mathbb{E}_{(x,y) \sim \gamma} |x - y| $$
Reinforcement Learning recasts molecular generation as a sequential decision problem. The policy gradient (REINFORCE) update is:
$$ \nabla J(\theta) = \mathbb{E}\left[G_{t} \frac{\nabla_{\theta} \pi_{\theta}(a_{t} \mid y_{1:t-1})}{\pi_{\theta}(a_{t} \mid y_{1:t-1})}\right] $$
To prevent RL fine-tuning from causing the generator to “drift” away from viable chemical structures, an augmented reward function incorporates the prior likelihood:
$$ R’(S) = [\sigma R(S) + \log P_{\text{prior}}(S) - \log P_{\text{current}}(S)]^{2} $$
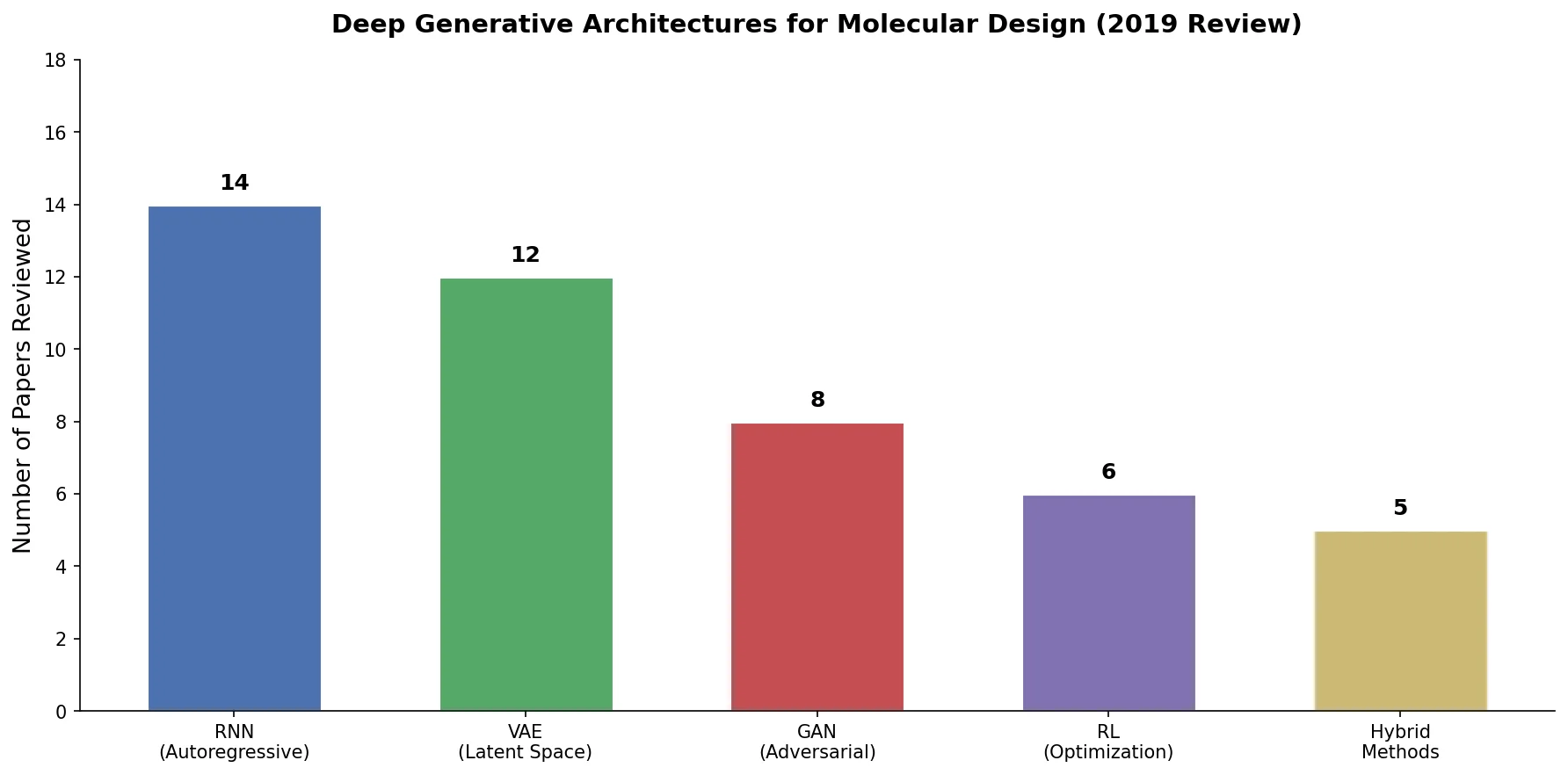
Cataloging 45 Models and Their Design Choices
Rather than running new experiments, the review’s methodology involves systematically cataloging and comparing 45 published models. Table 2 in the paper lists each model’s architecture, representation, training dataset, and dataset size. Key patterns include:
- RNN-based models (16 entries): Almost exclusively use SMILES, trained on ZINC or ChEMBL datasets with 0.1M-1.7M molecules.
- VAE variants (20 entries): The most diverse category, spanning SMILES VAEs, grammar VAEs, junction tree VAEs, graph-based VAEs, and 3D VAEs. Training sets range from 10K to 72M molecules.
- GAN models (7 entries): Include ORGAN, RANC, ATNC, MolGAN, and CycleGAN approaches. Notably, GANs appear to work with fewer training samples.
- Other approaches (2 entries): Pure RL methods from Zhou et al. and Stahl et al. that do not require pretraining on a dataset.
The review also catalogs 13 publicly available datasets (Table 3), ranging from QM9 (133K molecules with quantum chemical properties) to GDB-13 (977M combinatorially generated molecules) and ZINC15 (750M+ commercially available compounds).
Metrics and Reward Function Design
A significant contribution is the systematic treatment of reward functions. The review argues that generated molecules should satisfy six desiderata: diversity, novelty, stability, synthesizability, non-triviality, and good properties. Key metrics formalized include:
Diversity using Tanimoto similarity over fingerprints:
$$ r_{\text{diversity}} = 1 - \frac{1}{|\mathcal{G}|} \sum_{(x_{1}, x_{2}) \in \mathcal{G} \times \mathcal{G}} D(x_{1}, x_{2}) $$
Novelty measured as the fraction of generated molecules not appearing in a hold-out test set:
$$ r_{\text{novel}} = 1 - \frac{|\mathcal{G} \cap \mathcal{T}|}{|\mathcal{T}|} $$
Synthesizability primarily assessed via the SA score, sometimes augmented with ring penalties and medicinal chemistry filters.
The review also discusses the Fréchet ChemNet Distance as an analog of FID for molecular generation, and notes the emergence of standardized benchmarking platforms including MOSES, GuacaMol, and DiversityNet.
Key Findings and Future Directions
The review identifies several major trends and conclusions:
Shift from SMILES to graph-based representations. SMILES-based methods struggle with validity (the molecular autoencoder VAE achieved only 0.7-75% valid SMILES depending on sampling strategy). Methods that work directly on molecular graphs with chemistry-preserving operations achieve 100% validity, and the review predicts this trend will continue.
Advantages of adversarial and RL training over MLE. The mathematical analysis shows that MLE only optimizes forward KL divergence, which can lead to models that place probability mass where the data distribution is zero. GAN training optimizes the Jensen-Shannon divergence, which balances forward and reverse KL terms. RL approaches, particularly pure RL without pretraining, showed competitive performance with much less training data.
Genetic algorithms remain competitive. The review notes that the latest genetic algorithm approaches (Grammatical Evolution) could match deep learning methods for molecular optimization under some metrics, and at 100x lower computational cost in some comparisons. This serves as an important baseline calibration.
Reward function design is underappreciated. Early models generated unstable molecules with labile groups (enamines, hemiaminals, enol ethers). Better reward functions that incorporate synthesizability, diversity, and stability constraints significantly improved practical utility.
Need for standardized benchmarks. The review identifies a lack of agreement on evaluation methodology as a major barrier to progress, noting that published comparisons are often subtly biased toward novel methods.
Limitations
As a review paper from early 2019, the work predates several important developments: transformer-based architectures (which would soon dominate), SELFIES representations, diffusion models for molecules, and large-scale pretrained chemical language models. The review focuses primarily on drug-like small molecules and does not deeply cover protein design or materials optimization.
Reproducibility Details
Data
This is a review paper that does not present new experimental results. The paper catalogs 13 publicly available datasets used across the reviewed works:
| Purpose | Dataset | Size | Notes |
|---|---|---|---|
| Training/Eval | GDB-13 | 977M | Combinatorially generated library |
| Training/Eval | ZINC15 | 750M+ | Commercially available compounds |
| Training/Eval | GDB-17 | 50M | Combinatorially generated library |
| Training/Eval | ChEMBL | 2M | Curated bioactive molecules |
| Training/Eval | QM9 | 133,885 | Small organic molecules with DFT properties |
| Training/Eval | PubChemQC | 3.98M | PubChem compounds with DFT data |
Algorithms
The review provides mathematical derivations for MLE training (Eq. 1), VAE ELBO (Eqs. 9-13), AAE objectives (Eqs. 15-16), GAN objectives (Eqs. 19-22), WGAN (Eq. 24), REINFORCE gradient (Eq. 7), and numerous reward function formulations (Eqs. 26-36).
Evaluation
Key evaluation frameworks discussed:
- Fréchet ChemNet Distance (molecular analog of FID)
- MOSES benchmarking platform
- GuacaMol benchmarking suite
- Validity rate, uniqueness, novelty, and internal diversity metrics
Paper Information
Citation: Elton, D. C., Boukouvalas, Z., Fuge, M. D., & Chung, P. W. (2019). Deep Learning for Molecular Design: A Review of the State of the Art. Molecular Systems Design & Engineering, 4(4), 828-849. https://doi.org/10.1039/C9ME00039A
@article{elton2019deep,
title={Deep Learning for Molecular Design -- A Review of the State of the Art},
author={Elton, Daniel C. and Boukouvalas, Zois and Fuge, Mark D. and Chung, Peter W.},
journal={Molecular Systems Design \& Engineering},
volume={4},
number={4},
pages={828--849},
year={2019},
publisher={Royal Society of Chemistry},
doi={10.1039/C9ME00039A}
}