A Systematization of Chemical Language Models for Drug Design
This paper is a Systematization (minireview) that surveys the landscape of chemical language models (CLMs) for de novo drug design. It organizes the field along three axes: molecular string representations, deep learning architectures, and generation strategies (distribution learning, goal-directed, and conditional). The review also highlights experimental validations, current gaps, and future opportunities.
Why Chemical Language Models Matter for Drug Design
De novo drug design faces an enormous combinatorial challenge: the “chemical universe” is estimated to contain up to $10^{60}$ drug-like small molecules. Exhaustive enumeration is infeasible, and traditional design algorithms rely on hand-crafted assembly rules. Chemical language models address this by borrowing natural language processing techniques to learn the “chemical language,” generating molecules as string representations (SMILES, SELFIES, DeepSMILES) that satisfy both syntactic validity (chemically valid structures) and semantic correctness (desired pharmacological properties).
CLMs have gained traction because string representations are readily available for most molecular databases, generation is computationally cheap (one molecule per forward pass through a sequence model), and the same architecture can be applied to diverse tasks (property prediction, de novo generation, reaction prediction). At the time of this review, CLMs had produced experimentally validated bioactive molecules in several prospective studies, establishing them as practical tools for drug discovery.
Molecular String Representations: SMILES, DeepSMILES, and SELFIES
The review covers three main string representations used as input/output for CLMs:
SMILES (Simplified Molecular Input Line Entry Systems) converts hydrogen-depleted molecular graphs into strings where atoms are denoted by atomic symbols, bonds and branching by punctuation, and ring openings/closures by numbers. SMILES are non-univocal (multiple valid strings per molecule), and canonicalization algorithms are needed for unique representations. Multiple studies show that using randomized (non-canonical) SMILES for data augmentation improves CLM performance, with diminishing returns beyond 10- to 20-fold augmentation.
DeepSMILES modifies SMILES to improve machine-readability by replacing the paired ring-opening/closure digits with a count-based system and using closing parentheses only (no opening ones). This reduces the frequency of syntactically invalid strings but does not eliminate them entirely.
SELFIES (Self-Referencing Embedded Strings) use a formal grammar that guarantees 100% syntactic validity of decoded molecules. Every SELFIES string maps to a valid molecular graph. However, SELFIES can produce chemically unrealistic molecules (e.g., highly strained ring systems), and the mapping between string edits and molecular changes is less intuitive than for SMILES.
The review notes a key tradeoff: SMILES offer a richer, more interpretable language with well-studied augmentation strategies, while SELFIES guarantee validity at the cost of chemical realism and edit interpretability.
CLM Architectures and Training Strategies
Architectures
The review describes the main architectures used in CLMs:
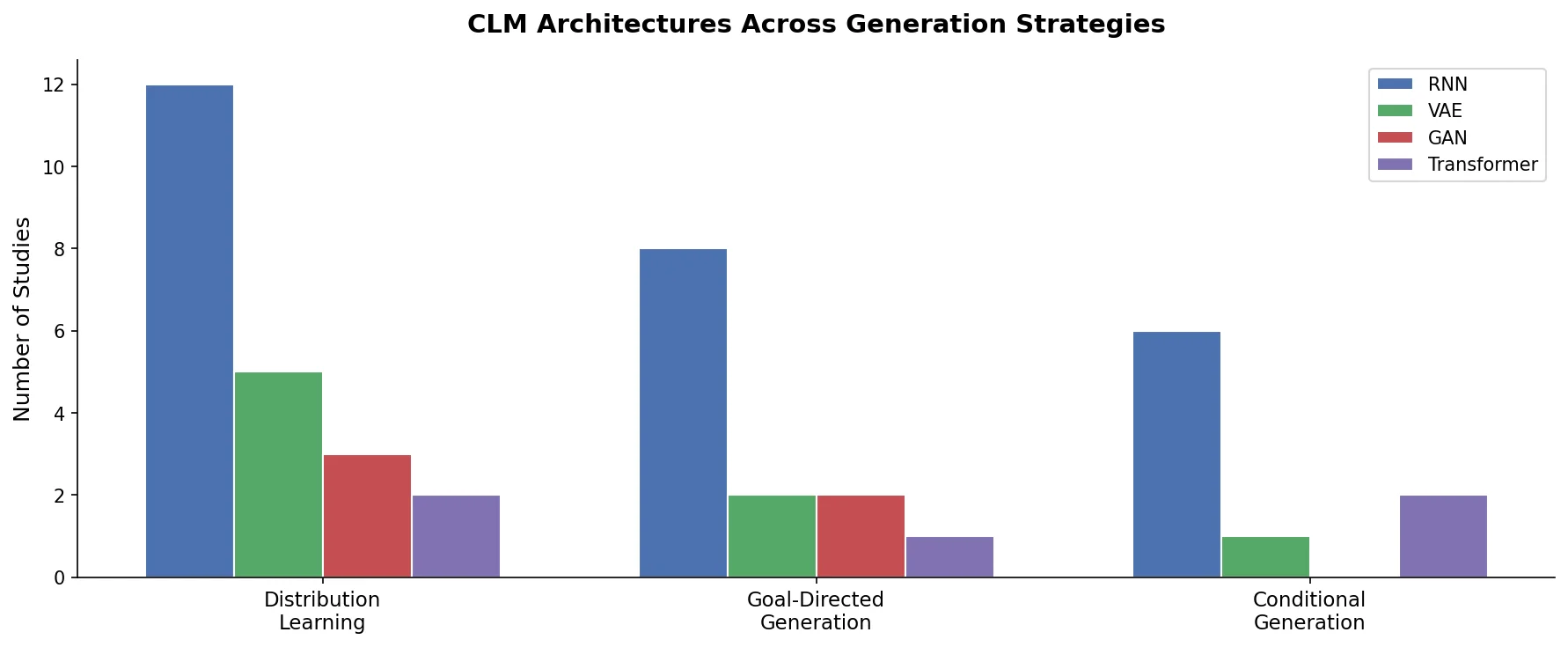
Recurrent Neural Networks (RNNs), particularly LSTMs and GRUs, dominated early CLM work. These models process SMILES character-by-character and generate new strings autoregressively via next-token prediction. RNNs are computationally efficient and well-suited to the sequential nature of molecular strings.
Variational Autoencoders (VAEs) encode molecules into a continuous latent space and decode them back into strings. This enables smooth interpolation between molecules and latent-space optimization, but generated strings may be syntactically invalid.
Generative Adversarial Networks (GANs) have been adapted for molecular string generation (e.g., ORGAN), though they face training instability and mode collapse challenges that limit their adoption.
Transformers have emerged as an increasingly popular alternative, offering parallelized training and the ability to capture long-range dependencies in molecular strings. The review notes the growing relevance of Transformer-based CLMs, particularly for large-scale pretraining.
Generation Strategies
The review organizes CLM generation into three categories:
Distribution learning: The model learns to reproduce the statistical distribution of a training set of molecules. No explicit scoring function is used during generation. The generated molecules are evaluated post-hoc by comparing their property distributions to the training set. This approach is end-to-end but provides no direct indication of individual molecule quality.
Goal-directed generation: A pretrained CLM is steered toward molecules optimizing a specified scoring function (e.g., predicted bioactivity, physicochemical properties). Common approaches include reinforcement learning (REINVENT and variants), hill-climbing, and Bayesian optimization. Scoring functions provide direct quality signals but can introduce biases, shortcuts, and limited structural diversity.
Conditional generation: An intermediate approach that learns a joint semantic space between molecular structures and desired properties. The desired property profile serves as an input “prompt” for generation (e.g., a protein target, gene expression signature, or 3D shape). This bypasses the need for external scoring functions but has seen limited experimental application.
Transfer Learning and Chemical Space Exploration
Transfer learning is the dominant paradigm for CLM-driven chemical space exploration. A large-scale pretraining step (on $10^5$ to $10^6$ molecules via next-character prediction) is followed by fine-tuning on a smaller set of molecules with desired properties (often 10 to $10^2$ molecules). Key findings from the literature:
- The minimum training set size depends on target molecule complexity and heterogeneity.
- SMILES augmentation is most beneficial with small training sets (fewer than 10,000 molecules) and plateaus for large, structurally complex datasets.
- Fine-tuning with as few as 10 to 100 molecules has produced experimentally validated bioactive designs.
- Hyperparameter tuning has relatively little effect on overall CLM performance.
Evaluating CLM Designs and Experimental Validation
The review identifies evaluation as a critical gap. CLMs are often benchmarked on “toy” properties such as calculated logP, molecular weight, or QED (quantitative estimate of drug-likeness). These metrics capture the ability to satisfy predefined criteria but fail to reflect real-world drug discovery complexity and may lead to trivial solutions.
Existing benchmarks (GuacaMol, MOSES) enable comparability across independently developed approaches but do not fully address the quality of generated compounds. The review emphasizes that experimental validation is the ultimate test. At the time of writing, only a few prospective applications had been published:
- Dual modulator of retinoid X and PPAR receptors (EC50 ranging from 0.06 to 2.3 uM)
- Inhibitor of Pim1 kinase and CDK4 (manually modified from generated design)
- Natural-product-inspired RORgamma agonist (EC50 = 0.68 uM)
- Molecules designed via combined generative AI and on-chip synthesis
The scarcity of experimental validations reflects the interdisciplinary expertise required and the time/cost of chemical synthesis.
Gaps, Limitations, and Future Directions
The review identifies several key gaps and opportunities:
Scoring function limitations: Current scoring functions struggle with activity cliffs and non-additive structure-activity relationships. Conditional generation methods may help overcome these limitations by learning direct structure-property mappings.
Structure-based design: Generating molecules that match electrostatic and shape features of protein binding pockets holds promise for addressing unexplored targets. However, prospective applications have been limited, potentially due to bias in existing protein-ligand affinity datasets.
Synthesizability: Improving the ability of CLMs to propose synthesizable molecules is expected to increase practical relevance. Automated synthesis platforms may help but could also limit accessible chemical space.
Few-shot learning: Large-scale pretrained CLMs combined with few-shot learning approaches are expected to boost prospective applications.
Extensions beyond small molecules: Extending chemical languages to more complex molecular entities (proteins with non-natural amino acids, crystals, supramolecular chemistry) is an open frontier.
Failure modes: Several studies have documented failure modes in goal-directed generation, including model shortcuts (exploiting scoring function artifacts), limited structural diversity, and generation of chemically unrealistic molecules.
Interdisciplinary collaboration: The review emphasizes that bridging deep learning, cheminformatics, and medicinal chemistry expertise is essential for translating CLM designs into real-world drug candidates.
Reproducibility Details
Data
This is a review paper and does not present novel experimental data. The paper surveys results from the literature.
Algorithms
No novel algorithms are introduced. The review categorizes existing approaches (RNNs, VAEs, GANs, Transformers) and generation strategies (distribution learning, goal-directed, conditional).
Models
No new models are presented. The paper references existing implementations including REINVENT, ORGAN, and various RNN-based and Transformer-based CLMs.
Evaluation
The review discusses existing benchmarks:
- GuacaMol: Benchmarking suite for de novo molecular design
- MOSES: Benchmarking platform for molecular generation models
- QED: Quantitative estimate of drug-likeness
- Various physicochemical property metrics (logP, molecular weight)
Hardware
Not applicable (review paper).
Paper Information
Citation: Grisoni, F. (2023). Chemical language models for de novo drug design: Challenges and opportunities. Current Opinion in Structural Biology, 79, 102527. https://doi.org/10.1016/j.sbi.2023.102527
Publication: Current Opinion in Structural Biology, Volume 79, April 2023
@article{grisoni2023chemical,
title={Chemical language models for de novo drug design: Challenges and opportunities},
author={Grisoni, Francesca},
journal={Current Opinion in Structural Biology},
volume={79},
pages={102527},
year={2023},
publisher={Elsevier},
doi={10.1016/j.sbi.2023.102527}
}