Paper Information
Citation: Yüksel, A., Ulusoy, E., Ünlü, A., & Doğan, T. (2023). SELFormer: Molecular Representation Learning via SELFIES Language Models. Machine Learning: Science and Technology, 4(2), 025035. https://doi.org/10.1088/2632-2153/acdb30
Publication: Machine Learning: Science and Technology 2023
Additional Resources:
A SELFIES-Based Chemical Language Model
This is primarily a Method paper ($\Psi_{\text{Method}}$) with a secondary Resource component ($\Psi_{\text{Resource}}$).
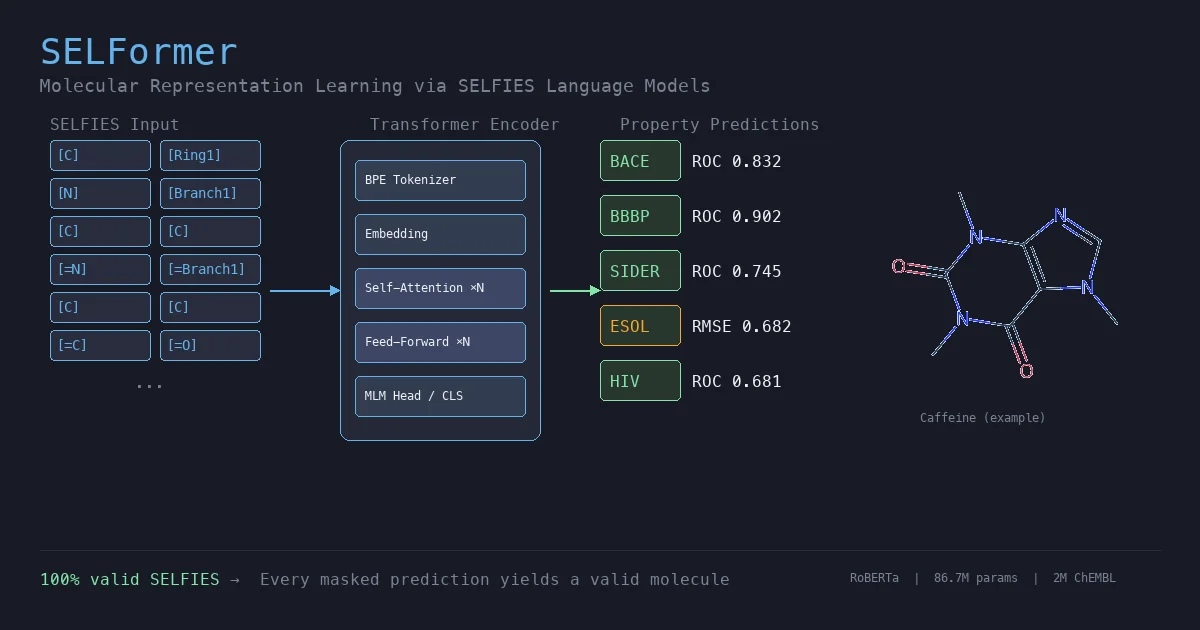
SELFormer applies the RoBERTa transformer architecture to SELFIES molecular string representations instead of the SMILES notation used by prior chemical language models. The model is pretrained via masked language modeling (MLM) on 2M drug-like compounds from ChEMBL and fine-tuned for molecular property prediction tasks on MoleculeNet benchmarks. The authors release pretrained models, fine-tuning code, and datasets as open-source resources.
Why SELFIES Over SMILES for Pretraining?
Existing chemical language models, including ChemBERTa, ChemBERTa-2, MolBERT, and MolFormer, all use SMILES as their input representation. SMILES has well-documented validity and robustness issues: arbitrary perturbations to a SMILES string frequently produce syntactically invalid outputs. This means a pretrained model must spend capacity learning SMILES grammar rules rather than chemical semantics.
SELFIES addresses this by construction: every possible SELFIES string decodes to a valid molecule. Despite this theoretical advantage and SELFIES’ growing adoption in generative chemistry, no prior work had systematically evaluated SELFIES as input for large-scale transformer pretraining. SELFormer fills this gap by providing a direct comparison between SELFIES-based and SMILES-based chemical language models on standard benchmarks.
Masked Language Modeling on Guaranteed-Valid Molecular Strings
SELFormer uses byte-level Byte-Pair Encoding (BPE) to tokenize SELFIES strings, then pretrains a RoBERTa encoder using the standard MLM objective. 15% of input tokens are masked, and the model minimizes the cross-entropy loss over the masked positions:
$$ \mathcal{L}_{\text{MLM}} = -\frac{1}{|\mathcal{M}|} \sum_{i \in \mathcal{M}} \log P(x_i \mid x_{\setminus \mathcal{M}}; \theta) $$
where $\mathcal{M}$ is the set of masked token indices, $x_i$ is the true token at position $i$, $x_{\setminus \mathcal{M}}$ is the corrupted input context, and $\theta$ are the model parameters.
The key insight is that because SELFIES guarantees 100% validity, every masked token prediction corresponds to a valid molecular fragment. The model never wastes capacity predicting invalid chemistry. For fine-tuning, a two-layer classification or regression head is added on top of the encoder’s output embedding.
Two model sizes were trained. Notably, the larger SELFormer uses fewer attention heads (4) but more hidden layers (12) than SELFormer-Lite (12 heads, 8 layers). This counterintuitive configuration emerged from the authors’ hyperparameter search over ~100 models, where deeper architectures with fewer heads outperformed wider, shallower ones:
| Configuration | SELFormer-Lite | SELFormer |
|---|---|---|
| Attention Heads | 12 | 4 |
| Hidden Layers | 8 | 12 |
| Batch Size | 16 | 16 |
| Learning Rate | 5e-5 | 5e-5 |
| Weight Decay | 0.01 | 0.01 |
| Pretraining Epochs | 100 | 100 |
| Parameters | 58.3M | 86.7M |
Benchmarking Against SMILES Transformers and Graph Models
SELFormer was pretrained on 2.08M drug-like compounds from ChEMBL v30 (converted from SMILES to SELFIES), then fine-tuned on nine MoleculeNet tasks. All evaluations use scaffold splitting via the Chemprop library.
Classification tasks (ROC-AUC, scaffold split):
| Model | BACE | BBBP | HIV | Tox21 | SIDER |
|---|---|---|---|---|---|
| SELFormer | 0.832 | 0.902 | 0.681 | 0.653 | 0.745 |
| ChemBERTa-2 | 0.799 | 0.728 | 0.622 | - | - |
| MolBERT | 0.866 | 0.762 | 0.783 | - | - |
| D-MPNN | 0.809 | 0.710 | 0.771 | 0.759 | 0.570 |
| MolCLR | 0.890 | 0.736 | 0.806 | 0.787 | 0.652 |
| GEM | 0.856 | 0.724 | 0.806 | 0.781 | 0.672 |
| KPGT | 0.855 | 0.908 | - | 0.848 | 0.649 |
Regression tasks (RMSE, scaffold split, lower is better):
| Model | ESOL | FreeSolv | Lipophilicity | PDBbind |
|---|---|---|---|---|
| SELFormer | 0.682 | 2.797 | 0.735 | 1.488 |
| ChemBERTa-2 | - | - | 0.986 | - |
| D-MPNN | 1.050 | 2.082 | 0.683 | 1.397 |
| GEM | 0.798 | 1.877 | 0.660 | - |
| KPGT | 0.803 | 2.121 | 0.600 | - |
The ablation study compared SELFormer vs. SELFormer-Lite across pretrained-only, 25-epoch, and 50-epoch fine-tuning configurations on randomly split datasets. SELFormer consistently outperformed SELFormer-Lite, confirming the benefit of the deeper (12-layer) architecture.
Strong Classification Performance with Compact Pretraining
SELFormer’s strongest results come on classification tasks where molecular substructure matters:
- SIDER: Best overall ROC-AUC (0.745), outperforming the next best method (MolCLR at 0.652) by 9.3 percentage points. The authors attribute this to SELFIES’ ability to capture subtle structural differences relevant to drug side effects.
- BBBP: Second best (0.902), behind only KPGT (0.908). SELFormer scored 17.4 percentage points above ChemBERTa-2 (0.728) on this task.
- BACE/HIV vs. ChemBERTa-2: SELFormer outperformed ChemBERTa-2 by 3.3 points on BACE (0.832 vs 0.799), 17.4 on BBBP, and 5.9 on HIV (0.681 vs 0.622). Since both models use similar RoBERTa architectures, this comparison is suggestive of a SELFIES advantage, though differences in pretraining corpus (ChEMBL vs PubChem), corpus size, and training procedure confound a clean attribution to the input representation alone.
- ESOL regression: Best RMSE (0.682) vs GEM (0.798), a 14.5% relative improvement.
Limitations are also apparent:
- HIV and Tox21: SELFormer underperforms graph-based methods (MolCLR, GEM, KPGT) on these larger datasets. The authors attribute this to insufficient hyperparameter search given computational constraints.
- FreeSolv and Lipophilicity regression: D-MPNN and graph-based methods maintain an edge, suggesting that explicit 2D/3D structural inductive biases remain valuable for certain property types.
- Small pretraining corpus: At 2M molecules, SELFormer’s corpus is orders of magnitude smaller than MolFormer’s 1.1B. Despite this, SELFormer outperforms MolFormer on SIDER (0.745 vs 0.690), highlighting SELFIES’ representational advantage.
- Single-task ablation scope: Some architectural claims rest on limited task coverage, and broader benchmarking would strengthen the conclusions.
Reproducibility Details
Data
| Purpose | Dataset | Size | Notes |
|---|---|---|---|
| Pretraining | ChEMBL v30 | 2,084,725 compounds (2,084,472 after SELFIES conversion) | Drug-like bioactive small molecules |
| Classification | BACE | 1,513 | Beta-secretase 1 inhibitor binding |
| Classification | BBBP | 2,039 | Blood-brain barrier permeability |
| Classification | HIV | 41,127 | HIV replication inhibition |
| Classification | SIDER | 1,427 | Drug side effects (27 classes) |
| Classification | Tox21 | 7,831 | Toxicity (12 targets) |
| Regression | ESOL | 1,128 | Aqueous solubility |
| Regression | FreeSolv | 642 | Hydration free energy |
| Regression | Lipophilicity | 4,200 | Octanol/water distribution coefficient |
| Regression | PDBbind | 11,908 | Binding affinity |
Algorithms
- Pretraining objective: Masked language modeling (MLM), 15% token masking
- Tokenization: Byte-level Byte-Pair Encoding (BPE) on SELFIES strings
- SMILES to SELFIES conversion: SELFIES API with Pandaral.lel for parallelization
- Splitting: Scaffold splitting via Chemprop library (80/10/10 train/validation/test)
- Fine-tuning: Two-layer classification/regression head on encoder output; up to 200 epochs with hyperparameter search
Models
- Architecture: RoBERTa (HuggingFace Transformers)
- SELFormer: 12 hidden layers, 4 attention heads, 86.7M parameters
- SELFormer-Lite: 8 hidden layers, 12 attention heads, 58.3M parameters
- Hyperparameter search: Sequential search over ~100 configurations on 100K molecule subset
Evaluation
| Metric | Task Type | Details |
|---|---|---|
| ROC-AUC | Classification | Area under receiver operating characteristic curve |
| PRC-AUC | Classification | Area under precision-recall curve (reported for random splits) |
| RMSE | Regression | Root mean squared error |
Results reported on scaffold split and random split datasets.
Hardware
- Compute: 2x NVIDIA A5000 GPUs
- Hyperparameter optimization time: ~11 days
- Full pretraining: 100 epochs on 2.08M molecules
Artifacts
| Artifact | Type | License | Notes |
|---|---|---|---|
| SELFormer GitHub | Code | GPL-3.0 | Pretraining, fine-tuning, and evaluation scripts |
| SELFormer on HuggingFace | Model | GPL-3.0 | Pretrained SELFormer weights |
| ChEMBL v30 | Dataset | CC BY-SA 3.0 | Source pretraining data |
| MoleculeNet | Benchmark | Unknown | Downstream evaluation tasks |
Citation
@article{yuksel2023selformer,
title={{SELFormer}: Molecular Representation Learning via {SELFIES} Language Models},
author={Y{\"u}ksel, Atakan and Ulusoy, Erva and {\"U}nl{\"u}, Atabey and Do{\u{g}}an, Tunca},
journal={Machine Learning: Science and Technology},
volume={4},
number={2},
pages={025035},
year={2023},
publisher={IOP Publishing},
doi={10.1088/2632-2153/acdb30}
}