Paper Information
Citation: Wang, Z., Nie, W., Qiao, Z., Xiao, C., Baraniuk, R. G., & Anandkumar, A. (2023). Retrieval-based Controllable Molecule Generation. Proceedings of the Eleventh International Conference on Learning Representations (ICLR 2023).
Publication: International Conference on Learning Representations (ICLR) 2023
Additional Resources:
@inproceedings{wang2023retrieval,
title={Retrieval-based Controllable Molecule Generation},
author={Wang, Zichao and Nie, Weili and Qiao, Zhuoran and Xiao, Chaowei and Baraniuk, Richard G. and Anandkumar, Anima},
booktitle={International Conference on Learning Representations},
year={2023},
url={https://openreview.net/forum?id=vDFA1tpuLvk}
}
Retrieval-Augmented Generation for Molecules
This is a Method paper that introduces RetMol, a retrieval-based framework for controllable molecule generation. The key idea is to guide a pre-trained generative model using a small set of exemplar molecules that partially satisfy the desired design criteria, retrieved from a task-specific database. The approach requires no task-specific fine-tuning of the generative backbone and works effectively with very few exemplar molecules (as few as 23).
Limitations of Existing Controllable Generation
Existing approaches to controllable molecule generation fall into three categories, each with drawbacks:
- Reinforcement learning (RL)-based methods require task-specific fine-tuning of the generative model for each new objective
- Supervised learning (SL)-based methods need molecules with desired properties as training data, which may be scarce
- Latent optimization-based methods require training property predictors in the latent space, which is challenging with limited active molecules and incompatible with variable-length latent spaces like those in transformers
RetMol addresses all three issues by keeping the generative backbone frozen and using a lightweight, task-agnostic retrieval module that can be applied to new tasks simply by swapping the retrieval database.
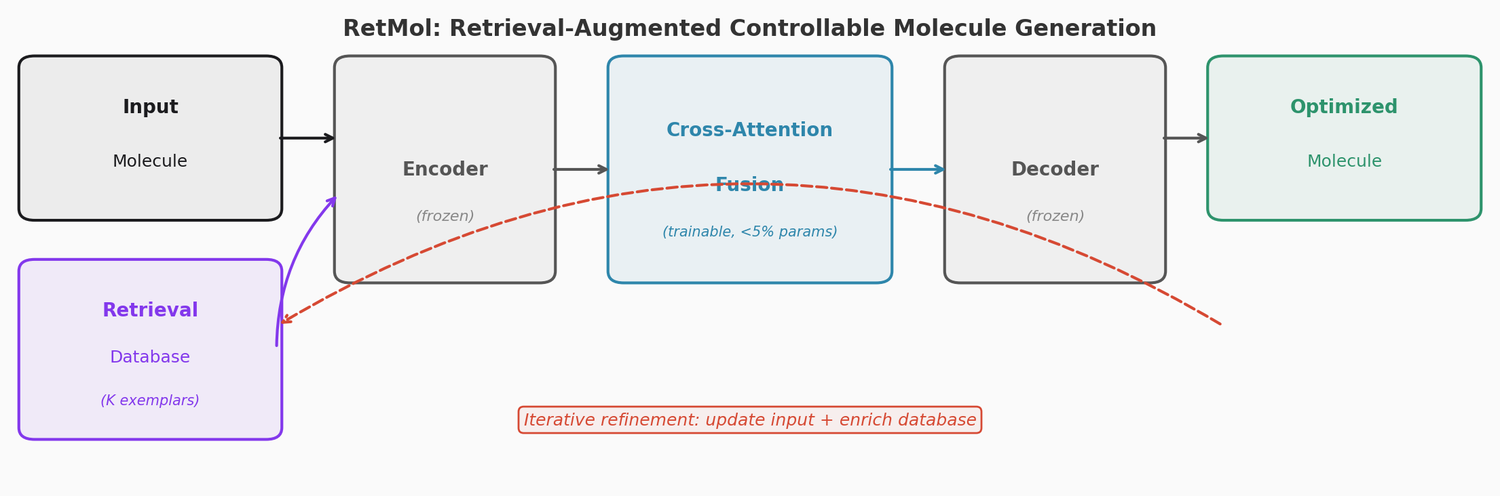
The RetMol Framework
RetMol consists of four components built around a pre-trained encoder-decoder backbone (Chemformer, a BART variant trained on ZINC):
Retrieval Database
A task-specific collection of exemplar molecules that at least partially satisfy the design criteria. The database can be very small (e.g., 23 known inhibitors for the SARS-CoV-2 task) and is dynamically updated during inference with newly generated molecules.
Molecule Retriever
A heuristic-based module that selects the $K$ most relevant exemplar molecules (default $K = 10$). It first constructs a feasible set of molecules satisfying all constraints, then selects those with the best property scores. If too few molecules satisfy all constraints, it progressively relaxes constraints until enough candidates are available.
Information Fusion via Cross-Attention
The core trainable component. Retrieved exemplar embeddings are fused with the input molecule embedding using cross-attention:
$$\boldsymbol{e} = f_{\text{CA}}(\boldsymbol{e}_{\text{in}}, \boldsymbol{E}_r; \theta) = \text{Attn}(\text{Query}(\boldsymbol{e}_{\text{in}}), \text{Key}(\boldsymbol{E}_r)) \cdot \text{Value}(\boldsymbol{E}_r)$$
where $\boldsymbol{e}_{\text{in}} = \text{Enc}(x_{\text{in}}) \in \mathbb{R}^{L \times D}$ is the input embedding and $\boldsymbol{E}_r = [\boldsymbol{e}_r^1, \ldots, \boldsymbol{e}_r^K]$ are the retrieved exemplar embeddings. This module adds less than 5% parameter overhead (460K parameters over the 10M base model).
Self-Supervised Training: Nearest Neighbor Prediction
Rather than reconstructing the input molecule (which would make the retrieval module unnecessary), RetMol trains the fusion module to predict the nearest neighbor of the input:
$$\mathcal{L}(\theta) = \sum_{i=1}^{B} \text{CE}\left(\text{Dec}\left(f_{\text{CA}}(\boldsymbol{e}_{\text{in}}^{(i)}, \boldsymbol{E}_r^{(i)}; \theta)\right), x_{\text{1NN}}^{(i)}\right)$$
The remaining $K - 1$ nearest neighbors serve as the retrieved exemplar molecules. This forces the fusion module to learn how to use exemplar molecules to transform the input toward a related target. Only the fusion module parameters are updated; the encoder and decoder remain frozen.
Iterative Refinement at Inference
During inference, RetMol uses an iterative process:
- Encode the input molecule and retrieved exemplars
- Fuse embeddings via cross-attention
- Perturb the fused embedding $M$ times with Gaussian noise
- Greedily decode $M$ candidate molecules
- Replace the input with the best candidate if it improves upon the current score
- Add remaining good candidates to the retrieval database
- Repeat until convergence or a maximum number of iterations
The dynamic update of the retrieval database is critical for extrapolating beyond the initial set of exemplar molecules.
Experiments and Results
RetMol is evaluated on four tasks of increasing difficulty:
QED Optimization Under Similarity Constraint
Goal: generate molecules with QED $\geq$ 0.9 while maintaining Tanimoto similarity $\geq$ 0.4 to the input. RetMol achieves 94.5% success rate, compared to 92.8% for the previous best (QMO).
Penalized LogP Optimization
Goal: maximize penalized LogP while maintaining structural similarity. At $\delta = 0.4$, RetMol achieves 11.55 average improvement, compared to 7.71 for QMO.
GSK3$\beta$ + JNK3 Dual Inhibitor Design
Goal: simultaneously satisfy four constraints (GSK3$\beta$ inhibition $\geq$ 0.5, JNK3 inhibition $\geq$ 0.5, QED $\geq$ 0.6, SA $\leq$ 4). Results:
| Method | Success % | Novelty | Diversity |
|---|---|---|---|
| REINVENT | 47.9 | 0.561 | 0.621 |
| RationaleRL | 74.8 | 0.568 | 0.701 |
| MARS | 92.3 | 0.824 | 0.719 |
| MolEvol | 93.0 | 0.757 | 0.681 |
| RetMol | 96.9 | 0.862 | 0.732 |
RetMol achieves this without task-specific fine-tuning and requires only 80 iterations compared to MARS’s 550.
SARS-CoV-2 Main Protease Inhibitor Optimization
A real-world task using only 23 known inhibitors as the retrieval database and optimizing 8 weakly-binding drugs. Under the milder similarity constraint ($\delta = 0.4$), RetMol achieves 2.84 kcal/mol average binding affinity improvement versus 1.67 for Graph GA. Under the stricter constraint ($\delta = 0.6$), RetMol succeeds on 5/8 molecules versus 3/8 for Graph GA.
Key Analysis Findings
- Database size: Strong performance even with 100 molecules, already outperforming baselines on success rate
- Database quality: Molecules satisfying all four constraints give the best results (96.9%), but partial satisfaction still works reasonably (84.7% with two properties)
- Training objective: The nearest neighbor prediction objective outperforms conventional reconstruction on validity (0.902 vs. 0.834) and uniqueness (0.922 vs. 0.665)
- Dynamic database update: Essential for extrapolating beyond the initial retrieval database, generating molecules with property values exceeding the best in the original database
Limitations
RetMol requires exemplar molecules that at least partially satisfy the design criteria. When such molecules are entirely unavailable, the framework cannot be applied. The method also relies on property predictors (for scoring and retrieval), whose accuracy directly affects generation quality. The iterative refinement process adds computational overhead at inference time, and the results depend on the Chemformer backbone’s generation capabilities.
Reproducibility
| Artifact | Type | License | Notes |
|---|---|---|---|
| NVlabs/RetMol | Code | NVIDIA Source Code License-NC | Full training and inference code |
| NVlabs/RetMol (checkpoints) | Model | CC BY-NC-SA 4.0 | Pre-trained model checkpoints |
Data: ZINC250k and ChEMBL datasets for training. Task-specific retrieval databases constructed from these datasets. COVID-19 task uses 23 known SARS-CoV-2 Mpro inhibitors.
Training: Information fusion module trained on 4x V100 GPUs (16GB each) for approximately 2 hours. Batch size of 256 per GPU, 50K iterations.
Inference: Single V100 GPU. Greedy decoding with Gaussian perturbation ($\sigma = 1$) for sampling multiple candidates per iteration.
Backbone: Chemformer (BART variant) pre-trained on ZINC. Frozen during RetMol training and inference.