A Method for Intrinsic Scoring and Bias Detection in Chemical Language Models
This is a Method paper that introduces two contributions to the chemical language model (CLM) pipeline for de novo molecular design. First, the authors propose using perplexity as a model-intrinsic score to rank generated SMILES strings by how well they match the design objectives encoded in the fine-tuning data. Second, they introduce a “delta score” that compares molecule rankings from pretrained and fine-tuned CLMs to detect pretraining bias, where molecules are generated primarily based on generic pretraining knowledge rather than task-specific fine-tuning objectives.
The Ranking and Bias Problem in CLM-Based Molecule Generation
Chemical language models generate new molecules as SMILES strings by iteratively predicting the next character based on learned probability distributions. After training, CLMs can produce large virtual libraries of candidate molecules via multinomial sampling. However, two key challenges remain: (1) the generated molecules lack a natural ranking, requiring external scoring methods such as similarity assessment or activity prediction for prioritization, and (2) transfer learning (pretraining on a large corpus followed by fine-tuning on a small target set) can introduce “pretraining bias,” where some generated molecules reflect generic chemical knowledge from pretraining rather than the specific design objectives of the fine-tuning data.
Beam search offers an alternative sampling approach that produces inherently ranked molecules by greedily selecting the most probable SMILES strings. However, beam search explores only a narrow portion of chemical space. The authors sought to combine the ranking advantage of beam search with the chemical space exploration of multinomial sampling by applying perplexity scoring as a post-hoc ranking criterion.
Perplexity Scoring and the Delta Score for Bias Estimation
The core innovation is the application of perplexity, a standard evaluation metric from natural language processing, to score SMILES strings generated by CLMs. For a SMILES string of length $N$ with character probabilities $p_i$ assigned by the CLM, perplexity is computed as:
$$ \text{perplexity} = 2^{-\frac{1}{N} \sum_{i=1}^{N} \log(p_{i})} $$
Low perplexity indicates that the CLM assigns high probability to each character in the SMILES string, suggesting the molecule closely matches the learned distribution of the fine-tuning data. The metric is normalized by string length, making it comparable across molecules of different sizes.
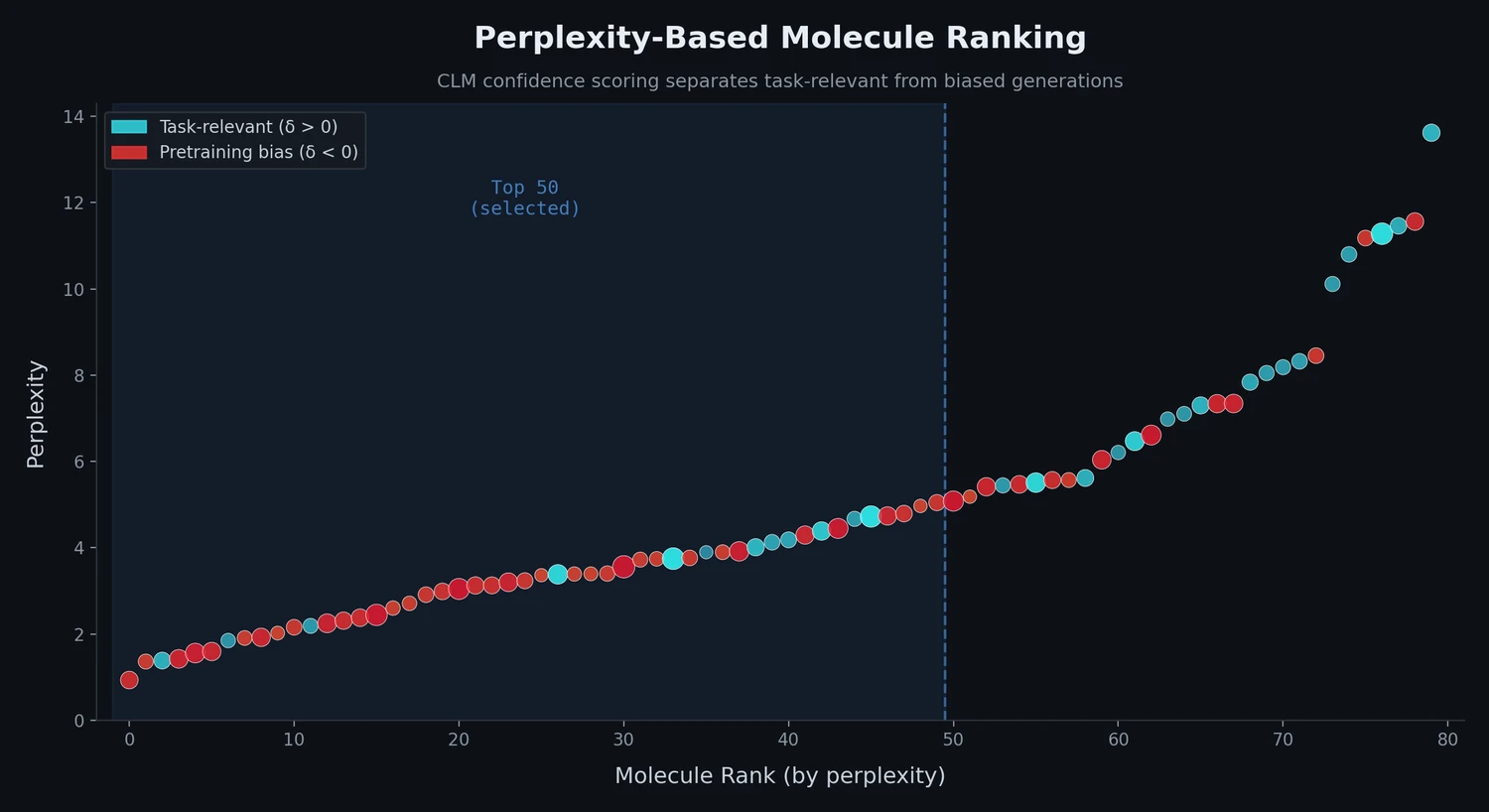
To address pretraining bias, the authors introduce a delta score. For each generated molecule, the perplexity-based rank from the fine-tuned model ($\text{rank}_{ft}$) is compared against the rank from the pretrained model ($\text{rank}_{pt}$):
$$ \text{delta} = \text{rank}_{ft} - \text{rank}_{pt} $$
A positive delta score indicates that the fine-tuned model ranks the molecule higher than the pretrained model, suggesting the molecule was generated based on task-specific fine-tuning knowledge. A negative delta score flags molecules that may have been generated primarily from pretraining information, which do not necessarily match the design objectives.
The multinomial sampling probability for each character is computed via the softmax function:
$$ p_{i} = \frac{e^{z_{i}/T}}{\sum_{j} e^{z_{j}/T}} $$
where $z_{i}$ is the CLM output logit for the $i$th character, $j$ runs over all dictionary characters, and $T$ is the temperature parameter (set to $T = 1$ in this study).
Experimental Setup: 10 Protein Targets Across Four Data Regimes
The authors systematically evaluated perplexity scoring across 10 macromolecular targets and four low-data fine-tuning regimes (5, 10, 20, and 40 molecules per target).
Model architecture: A four-layer LSTM-based RNN (5,820,515 parameters) with batch normalization layers, LSTM layers of 1024 and 256 units, trained using the Adam optimizer with a learning rate of $10^{-4}$.
Pretraining: The model was pretrained on 1,683,181 molecules from ChEMBL (version 28), encoded as canonical SMILES (20-90 characters), for 90 epochs.
Fine-tuning: For each of 10 randomly selected protein targets (Table 1), bioactive ligands with pChEMBL > 6 were selected. Fine-tuning sets of 5, 10, 20, and 40 molecules were compiled for each target. Fine-tuning ran for 100 epochs, with 1,000 SMILES strings sampled every second epoch via multinomial sampling ($T = 1$).
| CHEMBL ID | Target | Protein Classification |
|---|---|---|
| CHEMBL1836 | Prostanoid EP4 receptor | G protein-coupled receptor |
| CHEMBL1945 | Melatonin receptor 1A | G protein-coupled receptor |
| CHEMBL1983 | Serotonin 1D (5-HT1D) receptor | Family A GPCR |
| CHEMBL202 | Dihydrofolate reductase | Oxidoreductase |
| CHEMBL3522 | Cytochrome P450 17A1 | Cytochrome P450 |
| CHEMBL4029 | Interleukin-8 receptor A | Family A GPCR |
| CHEMBL5073 | CaM kinase I delta | Kinase |
| CHEMBL5137 | Metabotropic glutamate receptor 2 | G protein-coupled receptor |
| CHEMBL5408 | Serine/threonine-protein kinase TBK1 | Kinase |
| CHEMBL5608 | NT-3 growth factor receptor | Kinase |
Sampling comparison: Beam search sampling was performed with beam widths $k = 10$ and $k = 50$ for comparison against multinomial sampling.
Molecular similarity: Tanimoto similarity was computed using Morgan fingerprints (radius 2, length 1024) and 2D pharmacophore fingerprints via RDKit (2019.03.2).
Key Findings: Multinomial Sampling Outperforms Beam Search
Perplexity correlates with molecular similarity. The Pearson correlation between perplexity and Tanimoto distance to the fine-tuning set stabilized at approximately 0.5 across all data regimes. This correlation emerged earlier with larger fine-tuning sets. The result confirms that perplexity captures both substructural and pharmacophore features while also incorporating additional CLM-learned information.
Multinomial sampling produces better-ranked molecules than beam search. With the smallest fine-tuning sets (5 molecules), the top 50 molecules from multinomial sampling consistently exhibited lower (better) perplexity values than beam search at $k = 10$ or $k = 50$. Increasing the beam width from 10 to 50 did not markedly improve beam search performance. For novel molecules (Tanimoto similarity below 50% to the nearest fine-tuning compound), multinomial sampling identified lower-perplexity molecules in 72% of cases with the smallest fine-tuning sets.
Perplexity scoring narrows the quality distribution. The top 50 molecules selected by perplexity from multinomial sampling spanned a narrower range of perplexity values compared to beam search, suggesting a more consistent pool of high-quality candidates for follow-up synthesis.
Pretraining bias is substantial. The delta score analysis revealed that more than 40% of sampled molecules had negative delta scores during the first 20 fine-tuning epochs, meaning they were ranked higher by the pretrained model than the fine-tuned model. This fraction remained above 10% even at the end of 100 fine-tuning epochs across all data regimes, confirming that 10-40% of generated molecules reflect “generic” pretraining rather than task-focused fine-tuning.
Perplexity alone partially mitigates bias. Among the top 50 molecules selected by perplexity from multinomial sampling, only up to 3% had negative delta scores, compared to 10-40% in the unfiltered population. This suggests that perplexity-based ranking already reduces pretraining bias, though the delta score provides additional filtering power.
SMILES validity remained high. Mean SMILES string validity consistently exceeded 90% across all fine-tuned models and fine-tuning epochs.
Limitations
The authors note several limitations and future directions. The study used a fixed temperature of $T = 1$ for multinomial sampling; combining perplexity with temperature tuning or SMILES augmentation remains unexplored. The evaluation focused on 10 protein targets, and broader validation across diverse target classes would strengthen the conclusions. The authors also suggest that combining CLMs with perplexity scoring could be applied to screen large collections of commercially available compounds, which has not yet been tested.
Reproducibility Details
Data
| Purpose | Dataset | Size | Notes |
|---|---|---|---|
| Pretraining | ChEMBL v28 | 1,683,181 molecules | Canonical SMILES, 20-90 characters, salts and duplicates removed |
| Validation | ChEMBL v28 (split) | 84,160 molecules | Random split from pretraining set |
| Fine-tuning | ChEMBL v28 (per target) | 5, 10, 20, or 40 molecules | pChEMBL > 6, 10 targets |
Algorithms
- LSTM-based CLM with character-level SMILES prediction
- Multinomial sampling at $T = 1$
- Beam search at $k = 10$ and $k = 50$
- Perplexity computed per Equation 1; delta score per Equation 2
- Adam optimizer, learning rate $10^{-4}$, 90 pretraining epochs, 100 fine-tuning epochs
Models
- 4-layer LSTM RNN: batch normalization, LSTM (1024 units), LSTM (256 units), batch normalization
- 5,820,515 parameters total
- One-hot encoded SMILES input
- Pretrained weights available in the GitHub repository
Evaluation
| Metric | Description | Notes |
|---|---|---|
| Perplexity | Model confidence in generated SMILES | Lower is better |
| Delta score | Rank difference between fine-tuned and pretrained models | Positive indicates task-relevant generation |
| Tanimoto similarity | Morgan and pharmacophore fingerprints | Compared to fine-tuning set |
| Pearson correlation | Perplexity vs. Tanimoto distance | Stabilizes at ~0.5 |
| SMILES validity | Fraction of valid SMILES strings | Consistently > 90% |
Hardware
Hardware specifications are not reported in the paper. The implementation uses Keras (v2.2.0) with TensorFlow GPU backend (v1.9.0).
Artifacts
| Artifact | Type | License | Notes |
|---|---|---|---|
| CLM_perplexity | Code | MIT | Framework, pretrained weights, and training data |
| Beam search implementation | Code | Unknown | Referenced beam search implementation |
Paper Information
Citation: Moret, M., Grisoni, F., Katzberger, P., & Schneider, G. (2022). Perplexity-Based Molecule Ranking and Bias Estimation of Chemical Language Models. Journal of Chemical Information and Modeling, 62(5), 1199-1206. https://doi.org/10.1021/acs.jcim.2c00079
Publication: Journal of Chemical Information and Modeling, 2022
Additional Resources:
Citation
@article{moret2022perplexity,
title={Perplexity-Based Molecule Ranking and Bias Estimation of Chemical Language Models},
author={Moret, Michael and Grisoni, Francesca and Katzberger, Paul and Schneider, Gisbert},
journal={Journal of Chemical Information and Modeling},
volume={62},
number={5},
pages={1199--1206},
year={2022},
publisher={American Chemical Society},
doi={10.1021/acs.jcim.2c00079}
}