Transfer Learning Meets Molecular Property Prediction
This is a Method paper that introduces MolPMoFiT (Molecular Prediction Model Fine-Tuning), a transfer learning approach for QSPR/QSAR modeling. The primary contribution is adapting the ULMFiT framework from NLP to molecular property prediction by treating SMILES strings as a chemical language. A general-purpose molecular structure prediction model (MSPM) is pre-trained on one million ChEMBL molecules via self-supervised next-token prediction, then fine-tuned for specific QSAR endpoints. The approach achieves competitive or superior results to graph neural networks and descriptor-based methods across four benchmark datasets, with particular benefits for small datasets.
The Small Data Problem in QSAR Modeling
Deep learning models for molecular property prediction typically require large labeled training sets to learn useful structural representations. While methods like graph convolutional neural networks and SMILES-based models have achieved strong results on well-studied endpoints, they must be trained from scratch for each new task. This presents a challenge for small chemical datasets with limited labeled data, which remain common in drug discovery for specialized endpoints like allosteric inhibition, renal clearance, and inhibitor residence times.
Transfer learning had already shown transformative impact in computer vision (ImageNet pre-training) and NLP (ELMo, BERT, ULMFiT). In chemistry, prior transfer learning efforts included ChemNet (supervised pre-training on computed descriptors), Mol2vec (unsupervised substructure embeddings), and pre-trained graph neural networks. However, a systematic application of the ULMFiT self-supervised pre-training pipeline to SMILES-based molecular models had not been explored. MolPMoFiT fills this gap by treating the vast corpus of unlabeled molecular structures as a self-supervised training signal, analogous to how language models learn from unlabeled text.
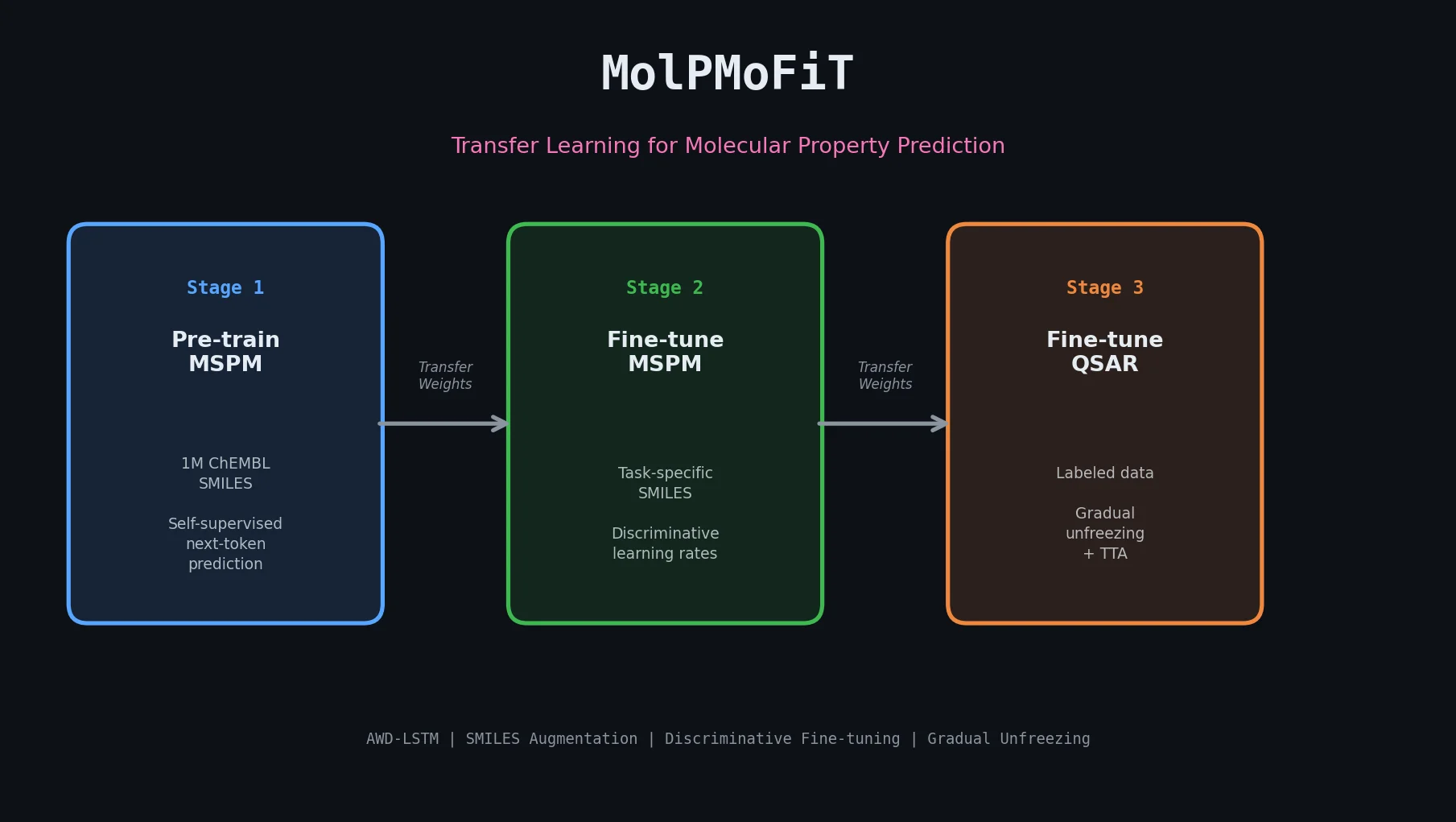
Core Innovation: ULMFiT Adapted for SMILES
MolPMoFiT adapts ULMFiT’s three-stage transfer learning pipeline to molecular property prediction:
Stage 1: General-Domain MSPM Pre-training. A molecular structure prediction model is trained on one million curated ChEMBL molecules to predict the next token in a SMILES string. This is purely self-supervised: the SMILES string provides its own labels. The model learns general chemical syntax and structural patterns.
Stage 2: Task-Specific MSPM Fine-tuning (Optional). The general MSPM is further fine-tuned on the unlabeled SMILES of the target task dataset. This adapts the language model to the specific chemical distribution of interest (e.g., HIV inhibitors vs. general bioactive molecules). Discriminative fine-tuning adjusts learning rates per layer:
$$\eta^{layer-1} = \eta^{layer} / 2.6$$
where higher layers (containing more task-specific features) receive higher learning rates.
Stage 3: QSAR/QSPR Model Fine-tuning. The embedding and encoder weights from the pre-trained MSPM are transferred to a new model with a task-specific classifier head. Fine-tuning uses three key techniques from ULMFiT:
- Discriminative fine-tuning: Different learning rates per layer group
- Gradual unfreezing: Layers are unfrozen sequentially (classifier first, then progressively deeper LSTM layers)
- One cycle policy: Learning rate scheduling following Smith’s approach
The model architecture is AWD-LSTM (ASGD Weight-Dropped LSTM) with an embedding dimension of 400, three LSTM layers with 1152 hidden units, and dropouts applied at every layer (embedding, input, weights, hidden). The QSAR classifier concatenates max pooling, mean pooling, and the last hidden state $h_T$ from the final LSTM layer, feeding this into two feedforward layers.
SMILES Augmentation. Since multiple valid SMILES can represent the same molecule through different atom orderings, the authors use SMILES enumeration as data augmentation. For regression tasks, Gaussian noise ($\sigma_{noise}$) is added to labels of augmented SMILES to simulate experimental error. Test-time augmentation (TTA) averages predictions across the canonical SMILES and four randomized SMILES.
Benchmarks Across Four QSAR Datasets
Datasets
| Dataset | Size | Task | Metric |
|---|---|---|---|
| Lipophilicity | 4,200 | Regression (logD) | RMSE |
| FreeSolv | 642 | Regression (solvation energy) | RMSE |
| HIV | 41,127 | Classification (replication inhibition) | AUROC |
| BBBP | 2,039 | Classification (blood-brain barrier) | AUROC |
All datasets use the same 10 random 80:10:10 splits from Yang et al. (2019) for fair comparison. Both random and scaffold splits were evaluated, with scaffold splits representing a more realistic test of generalization to novel chemical scaffolds.
Baselines
Models were compared against results reported by Yang et al. (2019): directed message passing neural network (D-MPNN), D-MPNN with RDKit features, random forest on Morgan fingerprints, feed-forward networks on Morgan fingerprints, and feed-forward networks on RDKit descriptors.
Hyperparameters
The same set of fine-tuning hyperparameters was used across all four tasks (tuned on the HIV dataset):
| Layer Group | Base Learning Rate | Epochs |
|---|---|---|
| Linear head only | 3e-2 | 4 |
| + Final LSTM layer | 5e-3 | 4 |
| + Final two LSTM layers | 5e-4 | 4 |
| Full model | 5e-5 | 6 |
Data augmentation settings were task-specific: lipophilicity training SMILES augmented 25x ($\sigma_{noise} = 0.3$); FreeSolv augmented 50x ($\sigma_{noise} = 0.5$); HIV active class augmented 60x and inactive 2x; BBBP positive class 10x and negative 30x.
Key Findings and Limitations
Benchmark Results
Lipophilicity (random split): MolPMoFiT achieved RMSE of $0.565 \pm 0.037$ with TTA and $0.625 \pm 0.032$ without, outperforming D-MPNN and other baselines.
FreeSolv (random split): RMSE of $1.197 \pm 0.127$ with TTA. The small dataset size (642 compounds) led to high variance across splits.
BBBP (random split): AUROC of $0.950 \pm 0.020$, outperforming all comparison models. Task-specific MSPM fine-tuning showed no clear benefit over the general MSPM.
HIV (random split): General MolPMoFiT achieved AUROC of $0.828 \pm 0.029$ with TTA. Task-specific fine-tuning yielded a slightly higher $0.834 \pm 0.025$ with TTA.
Scaffold splits consistently produced lower performance than random splits across all datasets, as expected for out-of-distribution generalization.
Transfer Learning Impact
Across all four datasets and varying training set sizes, MolPMoFiT consistently outperformed models trained from scratch with the same architecture. The improvement was most pronounced at smaller training set sizes, confirming the utility of pre-trained representations for low-data regimes.
SMILES Augmentation Analysis
Training data augmentation provided significant improvements across all tasks. For classification (HIV, BBBP), augmentation improved performance regardless of whether class re-balancing was applied. For regression (lipophilicity, FreeSolv), both SMILES augmentation and label noise were beneficial, with optimal noise levels varying by dataset.
Limitations
The authors note a fundamental limitation: the model learns mappings from individual SMILES strings to properties rather than from molecular structures to properties. SMILES augmentation acts as a regularization technique to mitigate this, making the model more robust to different SMILES representations of the same molecule. The task-specific MSPM fine-tuning stage did not consistently improve results, requiring further investigation. All hyperparameters were tuned on one dataset (HIV) and applied uniformly, which may not be optimal for all endpoints.
Reproducibility Details
Data
| Purpose | Dataset | Size | Notes |
|---|---|---|---|
| Pre-training | ChEMBL (curated) | 1M molecules | Filtered: no mixtures, max 50 heavy atoms, standardized with MolVS, canonized with RDKit |
| Evaluation | Lipophilicity | 4,200 | MoleculeNet benchmark |
| Evaluation | FreeSolv | 642 | MoleculeNet benchmark |
| Evaluation | HIV | 41,127 | MoleculeNet benchmark |
| Evaluation | BBBP | 2,039 | MoleculeNet benchmark |
Algorithms
- AWD-LSTM architecture with embedding dim 400, three LSTM layers (1152 hidden units), dropouts at all layers
- ULMFiT fine-tuning: discriminative learning rates ($\eta^{layer-1} = \eta^{layer}/2.6$), gradual unfreezing, one cycle policy
- SMILES character-level tokenization with special handling for two-character tokens (Cl, Br) and bracket-enclosed tokens
- SMILES enumeration for data augmentation with optional Gaussian label noise for regression
Models
- General-domain MSPM pre-trained on 1M ChEMBL molecules (10 epochs)
- Task-specific MSPMs fine-tuned per dataset (optional stage)
- QSAR models fine-tuned with transferred embeddings and encoder
Evaluation
| Dataset | Split | Metric | MolPMoFiT (TTA) | Best Baseline |
|---|---|---|---|---|
| Lipophilicity | Random | RMSE | $0.565 \pm 0.037$ | D-MPNN |
| Lipophilicity | Scaffold | RMSE | $0.635 \pm 0.031$ | D-MPNN |
| FreeSolv | Random | RMSE | $1.197 \pm 0.127$ | D-MPNN |
| FreeSolv | Scaffold | RMSE | $2.082 \pm 0.460$ | D-MPNN |
| BBBP | Random | AUROC | $0.950 \pm 0.020$ | D-MPNN |
| BBBP | Scaffold | AUROC | $0.931 \pm 0.025$ | D-MPNN |
| HIV | Random | AUROC | $0.828 \pm 0.029$ | D-MPNN |
| HIV | Scaffold | AUROC | $0.816 \pm 0.022$ | D-MPNN |
Hardware
- NVIDIA Quadro P4000 GPU (single GPU)
- General-domain MSPM pre-training: approximately 1 day
- Pre-training needs to be done only once; fine-tuning is fast per task
Artifacts
| Artifact | Type | License | Notes |
|---|---|---|---|
| MolPMoFiT | Code | Not specified | PyTorch + fastai v1 implementation with curated datasets |
Paper Information
Citation: Li, X., & Fourches, D. (2020). Inductive transfer learning for molecular activity prediction: Next-Gen QSAR Models with MolPMoFiT. Journal of Cheminformatics, 12, 27. https://doi.org/10.1186/s13321-020-00430-x
@article{li2020molpmofit,
title={Inductive transfer learning for molecular activity prediction: Next-Gen QSAR Models with MolPMoFiT},
author={Li, Xinhao and Fourches, Denis},
journal={Journal of Cheminformatics},
volume={12},
number={1},
pages={27},
year={2020},
doi={10.1186/s13321-020-00430-x}
}