A Graph-Aware BERT for Molecular Property Prediction
MG-BERT is a Method paper that adapts the BERT pretraining paradigm from NLP to molecular graphs. The primary contribution is a modified Transformer architecture that replaces global self-attention with bond-based local attention, allowing atoms to exchange information only through chemical bonds. This creates a deep message-passing network that avoids the oversmoothing problem of conventional graph neural networks (GNNs). Combined with a masked atom prediction pretraining strategy on 1.7 million unlabeled molecules from ChEMBL, MG-BERT learns context-sensitive atomic representations that transfer effectively to downstream property prediction tasks.
Data Scarcity in Molecular Property Prediction
Molecular property prediction is central to drug discovery, particularly for ADMET (Absorption, Distribution, Metabolism, Excretion, and Toxicity) endpoints. While deep learning has advanced many domains, molecular property prediction faces a persistent challenge: labeled data scarcity. ADMET measurements require expensive, time-consuming experiments, and typical datasets contain only hundreds to thousands of examples.
Prior approaches fall into three categories, each with limitations:
- Feature engineering (molecular fingerprints, descriptors): Requires expert design, suffers from low scalability, and fixed representations cannot be optimized for specific tasks.
- SMILES-based deep learning (CNNs, LSTMs, Transformers on SMILES strings): Must learn to parse molecular information from complex string syntax, increasing learning difficulty. Autoencoder-based methods (e.g., CDDD) learn fixed representations that cannot be fine-tuned.
- Graph neural networks (GAT, GCN): Can learn directly from molecular topology, but are limited to 2-3 layers due to oversmoothing, restricting their capacity to capture deep-level patterns.
The BERT model from NLP demonstrated that self-supervised pretraining on large unlabeled corpora followed by fine-tuning on small labeled datasets can substantially improve downstream performance. SMILES-BERT applied this idea to SMILES strings directly, but suffered from interpretability issues due to auxiliary characters in the SMILES syntax. MG-BERT addresses these limitations by operating directly on molecular graphs.
Bond-Based Local Attention and Masked Atom Pretraining
The core innovation of MG-BERT has two components: a modified Transformer architecture for molecular graphs and a self-supervised pretraining strategy.
Architecture Modifications
The original BERT model uses three components: an embedding layer, Transformer encoder layers, and a task-specific output layer. MG-BERT makes three key modifications:
Atom embeddings replace word embeddings. The dictionary contains 16 tokens: 13 common atom types ([H], [C], [N], [O], [F], [S], [Cl], [P], [Br], [B], [I], [Si], [Se]), plus [UNK] for rare atoms, [MASK] for pretraining, and [GLOBAL] for graph-level readout.
No positional encoding. Unlike sequential text, atoms in a molecular graph have no inherent ordering, so positional embeddings are removed.
Local attention replaces global attention. The adjacency matrix of the molecular graph is used as a visibility matrix to modulate the attention scores. Each atom can only attend to atoms connected by chemical bonds. Formally, the attention is constrained so that:
$$A’_{ij} = \begin{cases} A_{ij} & \text{if bond exists between } i \text{ and } j \\ -\infty & \text{otherwise} \end{cases}$$
where $A_{ij}$ is the standard scaled dot-product attention score. This local message passing makes MG-BERT a variant of GNN, but one that can stack many layers (6 in the medium configuration) without oversmoothing, thanks to the residual connections inherited from the Transformer architecture.
- Supernode for graph-level readout. A [GLOBAL] supernode is added to each molecular graph, connected to all atoms. This node aggregates information from the entire molecule and serves as the molecular representation for downstream prediction.
Masked Atom Prediction
The pretraining strategy mirrors BERT’s masked language model but operates on atoms:
- 15% of atoms in each molecule are randomly selected (at least one atom per molecule)
- Of selected atoms: 80% are replaced with [MASK], 10% are randomly replaced with another atom type, and 10% remain unchanged
- The model is trained to predict the original atom type at masked positions
- Loss is computed only at masked positions
Model Configurations
Three model sizes were compared:
| Configuration | Layers | Heads | Embedding Size | FFN Size | Recovery Accuracy |
|---|---|---|---|---|---|
| MG-BERT Small | 3 | 2 | 128 | 256 | 95.27% |
| MG-BERT Medium | 6 | 4 | 256 | 512 | 98.31% |
| MG-BERT Large | 12 | 8 | 576 | 1152 | 98.35% |
The medium configuration was selected for all experiments because it achieved the best downstream performance, despite the large model having slightly higher pretraining recovery accuracy. The authors attribute this to overfitting risk with the larger model.
Experimental Setup and Baselines
Pretraining
MG-BERT was pretrained on 1.7 million compounds randomly selected from ChEMBL, with 10% held out for evaluation (1.53M training molecules). Molecules were converted to 2D undirected graphs using RDKit, with hydrogen atoms explicitly included. The model was pretrained for 10 epochs using Adam with learning rate 1e-4 and batch size 256.
Fine-tuning Datasets
Sixteen datasets covering ADMET endpoints and common molecular properties were collected from ADMETlab and MoleculeNet:
| Type | Dataset | Category | Size |
|---|---|---|---|
| Regression | Caco2 | Absorption | 979 |
| Regression | logD | Physicochemical | 10,354 |
| Regression | logS | Physicochemical | 5,045 |
| Regression | PPB | Distribution | 1,480 |
| Regression | tox | Toxicity | 7,295 |
| Regression | ESOL | Physicochemical | 1,128 |
| Regression | FreeSolv | Physicochemical | 642 |
| Regression | Lipo | Physicochemical | 4,200 |
| Classification | Ames | Toxicity | 6,719 |
| Classification | BBB | Distribution | 1,855 |
| Classification | FDAMDD | Toxicity | 795 |
| Classification | H_HT | Toxicity | 2,170 |
| Classification | Pgp_inh | Absorption | 2,125 |
| Classification | Pgp_sub | Absorption | 1,210 |
| Classification | BACE | Biophysics | 1,513 |
| Classification | BBBP | Physiology | 2,039 |
Datasets were split 8:1:1 (train:validation:test) with stratified sampling by SMILES length. Each experiment was repeated 10 times with random splits, reporting mean and standard deviation. Regression was evaluated by R-squared, classification by ROC-AUC. Early stopping with a maximum of 100 epochs was used.
Baselines
Five baselines were compared:
- ECFP4-XGBoost: Extended connectivity fingerprints (diameter 4) with gradient-boosted trees
- GAT: Graph Attention Network
- GCN: Graph Convolutional Network
- CDDD: Continuous and Data-Driven Descriptors (pretrained RNN encoder on SMILES with a fully connected network)
- SMILES-BERT: Original BERT applied directly to SMILES strings
Ablation Studies
Two ablation studies were conducted:
- Pretraining effectiveness: Comparing pretrained vs. non-pretrained MG-BERT under identical hyperparameters
- Hydrogen atoms: Comparing MG-BERT with and without explicit hydrogen atoms in the molecular graph
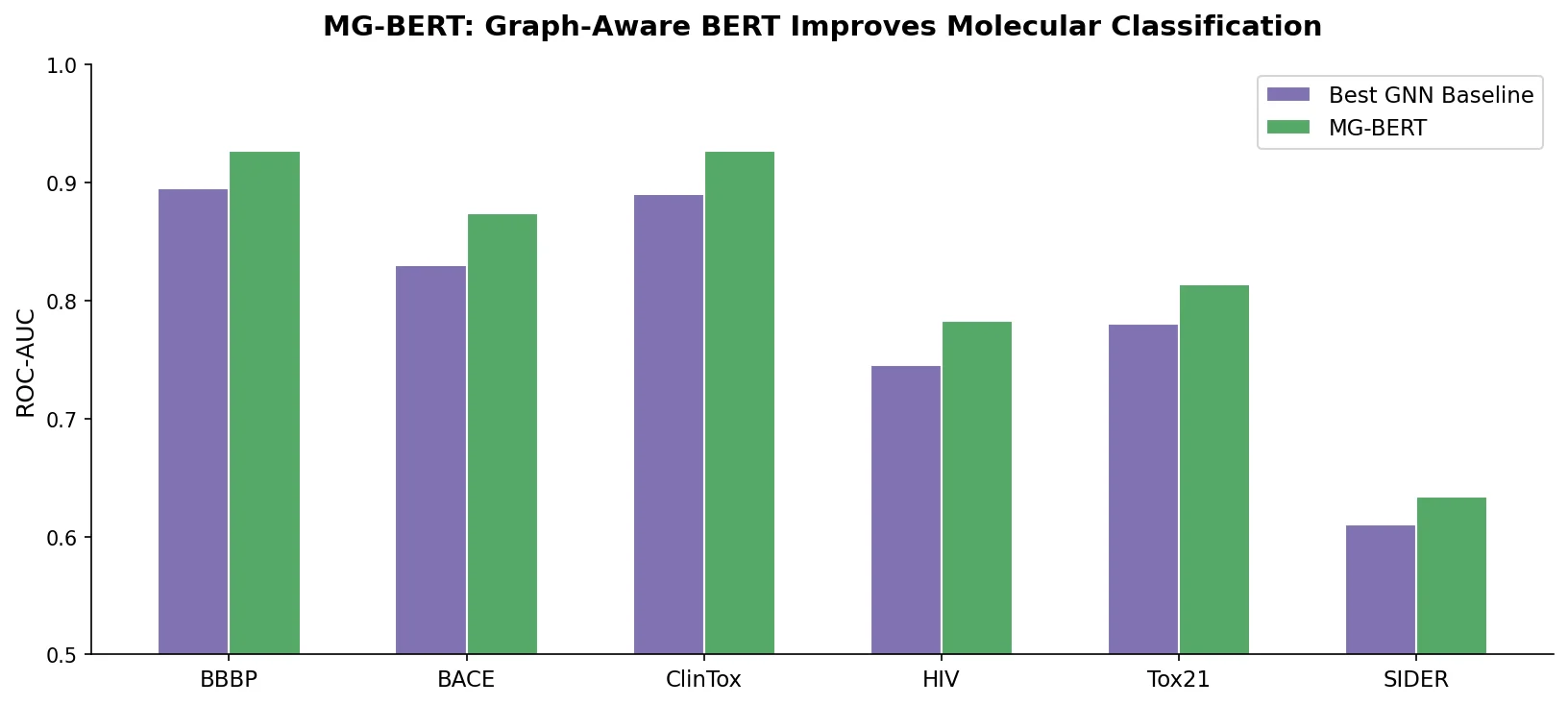
Consistent Improvements Across ADMET Benchmarks
Main Results
MG-BERT consistently outperformed all baselines across all 16 datasets. Key results on the 11 ADMET datasets:
| Dataset | ECFP4-XGBoost | GAT | GCN | CDDD | SMILES-BERT | MG-BERT |
|---|---|---|---|---|---|---|
| Caco2 (R2) | 61.41 | 69.16 | 67.15 | 73.42 | 72.39 | 74.68 |
| logD (R2) | 70.84 | 84.62 | 86.22 | 85.85 | 86.31 | 87.46 |
| logS (R2) | 73.73 | 84.06 | 83.47 | 84.01 | 85.20 | 87.66 |
| PPB (R2) | 55.11 | 59.96 | 57.34 | 54.12 | 62.37 | 65.94 |
| Ames (AUC) | 87.21 | 86.38 | 87.04 | 86.82 | 87.69 | 89.33 |
| BBB (AUC) | 94.62 | 93.03 | 92.67 | 94.44 | 94.02 | 95.41 |
| BBBP (AUC) | 89.16 | 90.33 | 90.74 | 91.12 | 91.32 | 92.08 |
The overall improvement across all datasets was 28.1% (7.02% on classification, 21.28% on regression). Improvements were statistically significant at the 95% confidence level (paired t-test, P <= 0.001).
Pretraining Ablation
Pretraining improved performance by more than 2% on all datasets. The benefit was largest for small datasets: Caco2 improved by approximately 10 percentage points (64.79 to 74.68 R2), and FDAMDD improved by about 7.5 points (80.76 to 88.23 AUC). This confirms that self-supervised pretraining effectively addresses the labeled data scarcity problem.
Hydrogen Atom Ablation
Including explicit hydrogen atoms improved pretraining recovery accuracy from 92.25% to 98.31% and consistently improved downstream performance. The authors provide an intuitive explanation: hydrogen atoms help determine bond counts for neighboring atoms, which is critical for the masked atom recovery task. They also show that removing hydrogens can make structurally distinct molecules (e.g., benzene and cyclohexane) indistinguishable at the graph level.
Interpretability via Attention Visualization
The authors provide two forms of interpretability analysis:
t-SNE visualization of atomic representations: Pretrained atomic representations cluster by atom type and, more specifically, by local chemical environment (e.g., aromatic carbons separate from aliphatic carbons, C-N bonds from C-O bonds). This demonstrates that pretraining captures neighborhood context beyond simple atom identity.
Attention weight visualization: On the logD task, the supernode’s attention focuses on polar groups (which govern lipophilicity). On the Ames mutagenicity task, attention concentrates on known mutagenic structural alerts (acylchloride, nitrosamide, azide groups). This provides chemically meaningful explanations for predictions.
Limitations
The paper does not extensively discuss limitations, but several can be identified:
- The model uses only 2D molecular topology (atom types and bonds) without 3D conformational information or bond-type features
- The atom dictionary is limited to 13 common types plus [UNK], which may lose information for molecules containing rarer elements
- Evaluation is limited to ADMET-focused datasets; broader chemical spaces (e.g., materials, catalysts) are not tested
- The comparison baselines do not include other graph-based pretraining methods (e.g., the contemporaneous Strategies for Pre-training Graph Neural Networks by Hu et al.)
Reproducibility Details
Data
| Purpose | Dataset | Size | Notes |
|---|---|---|---|
| Pretraining | ChEMBL (random subset) | 1.7M molecules (1.53M train) | 10% held out for evaluation |
| Fine-tuning | ADMETlab + MoleculeNet | 16 datasets (642-10,354 molecules) | 8:1:1 splits, stratified by SMILES length |
Algorithms
- Optimizer: Adam (pretraining: lr=1e-4, batch=256; fine-tuning: lr from {1e-5, 5e-5, 1e-4}, batch from {16, 32, 64})
- Pretraining epochs: 10
- Fine-tuning: Up to 100 epochs with early stopping
- Dropout: Optimized per task in range [0.0, 0.5]
- Masking: 15% of atoms (80% [MASK], 10% random, 10% unchanged)
Models
- Architecture: MG-BERT Medium (6 layers, 4 heads, embedding size 256, FFN size 512)
- Molecule processing: RDKit for graph conversion with explicit hydrogens
Evaluation
| Metric | Task Type | Notes |
|---|---|---|
| R-squared (R2) | Regression | Higher is better |
| ROC-AUC | Classification | Higher is better |
| Accuracy, RMSE | Both | Reported in supplementary Table S1 |
All results averaged over 10 random splits with standard deviations reported.
Hardware
The paper does not specify hardware requirements (GPU type, training time, or memory usage).
Artifacts
| Artifact | Type | License | Notes |
|---|---|---|---|
| Molecular-graph-BERT | Code | Not specified | Jupyter Notebook implementation; last code push August 2021 |
Paper Information
Citation: Zhang, X.-C., Wu, C.-K., Yang, Z.-J., Wu, Z.-X., Yi, J.-C., Hsieh, C.-Y., Hou, T.-J., & Cao, D.-S. (2021). MG-BERT: leveraging unsupervised atomic representation learning for molecular property prediction. Briefings in Bioinformatics, 22(6), bbab152. https://doi.org/10.1093/bib/bbab152
@article{zhang2021mgbert,
title={{MG-BERT}: leveraging unsupervised atomic representation learning for molecular property prediction},
author={Zhang, Xiao-Chen and Wu, Cheng-Kun and Yang, Zhi-Jiang and Wu, Zhen-Xing and Yi, Jia-Cai and Hsieh, Chang-Yu and Hou, Ting-Jun and Cao, Dong-Sheng},
journal={Briefings in Bioinformatics},
volume={22},
number={6},
pages={bbab152},
year={2021},
publisher={Oxford University Press},
doi={10.1093/bib/bbab152}
}