Paper Information
Citation: Du, Y., Fu, T., Sun, J., & Liu, S. (2022). MolGenSurvey: A Systematic Survey in Machine Learning Models for Molecule Design. arXiv preprint arXiv:2203.14500.
Publication: arXiv preprint, March 2022. Note: This survey covers literature through early 2022 and does not include subsequent advances in diffusion models, LLMs for chemistry, or flow matching.
Additional Resources:
@article{du2022molgensurvey,
title={MolGenSurvey: A Systematic Survey in Machine Learning Models for Molecule Design},
author={Du, Yuanqi and Fu, Tianfan and Sun, Jimeng and Liu, Shengchao},
journal={arXiv preprint arXiv:2203.14500},
year={2022}
}
A Taxonomy for ML-Driven Molecule Design
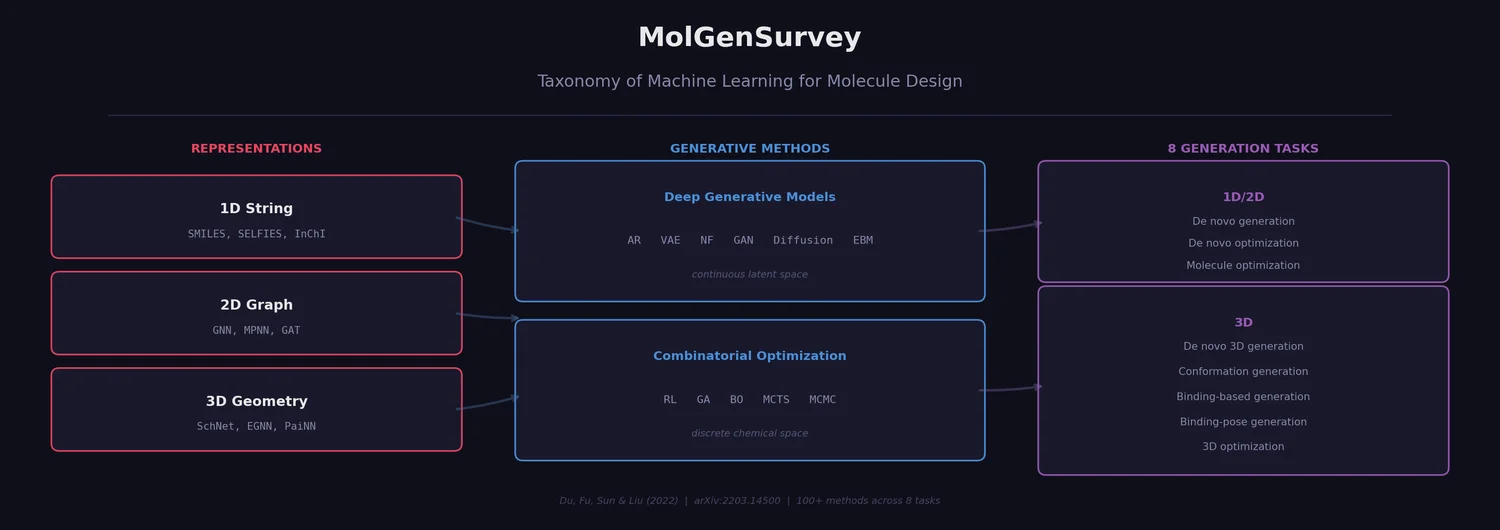
This is a Systematization paper that reviews machine learning approaches for molecule design across all three major molecular representations (1D string, 2D graph, 3D geometry) and both deep generative and combinatorial optimization paradigms. Prior surveys [8]-[14] each covered subsets of the literature (e.g., only generative methods, or only specific task types). MolGenSurvey extends these by unifying the field into a single taxonomy based on input type, output type, and generation goal, identifying eight distinct molecule generation tasks. It catalogs over 100 methods across these categories and provides a structured comparison of evaluation metrics, datasets, and experimental setups.
The chemical space of drug-like molecules is estimated at $10^{23}$ to $10^{60}$, making exhaustive enumeration computationally infeasible. Traditional high-throughput screening searches existing databases but is slow and expensive. ML-based generative approaches offer a way to intelligently explore this space, either by learning continuous latent representations (deep generative models) or by directly searching the discrete chemical space (combinatorial optimization methods).
Molecular Representations
The survey identifies three mainstream featurization approaches for molecules, each carrying different tradeoffs for generation tasks.
1D String Descriptions
SMILES and SELFIES are the two dominant string representations. SMILES encodes molecules as character strings following grammar rules for bonds, branches, and ring closures. Its main limitation is that arbitrary strings are often chemically invalid. SELFIES augments the encoding rules for branches and rings to achieve 100% validity by construction.
Other string representations exist (InChI, SMARTS) but are less commonly used for generation. Representation learning over strings has adopted CNNs, RNNs, and Transformers from NLP.
2D Molecular Graphs
Molecules naturally map to graphs where atoms are nodes and bonds are edges. Graph neural networks (GNNs), particularly those following the message-passing neural network (MPNN) framework, have become the standard representation method. The MPNN updates each node’s representation by aggregating information from its $K$-hop neighborhood. Notable architectures include D-MPNN (directional message passing), PNA (diverse aggregation methods), AttentiveFP (attention-based), and Graphormer (transformer-based).
3D Molecular Geometry
Molecules are inherently 3D objects with conformations (3D structures at local energy minima) that determine function. Representing 3D geometry requires models that respect E(3) or SE(3) equivariance (invariance to rotation and translation). The survey catalogs architectures along this line including SchNet, DimeNet, EGNN, SphereNet, and PaiNN.
Additional featurization methods (molecular fingerprints/descriptors, 3D density maps, 3D surface meshes, and chemical images) are noted but have seen limited use in generation tasks.
Deep Generative Models
The survey covers six families of deep generative models applied to molecule design.
Autoregressive Models (ARs)
ARs factorize the joint distribution of a molecule as a product of conditional distributions over its subcomponents:
$$p(\boldsymbol{x}) = \prod_{i=1}^{d} p(\bar{x}_i \mid \bar{x}_1, \bar{x}_2, \ldots, \bar{x}_{i-1})$$
For molecular graphs, this means sequentially predicting the next atom or bond conditioned on the partial structure built so far. RNNs, Transformers, and BERT-style models all implement this paradigm.
Variational Autoencoders (VAEs)
VAEs learn a continuous latent space by maximizing the evidence lower bound (ELBO):
$$\log p(\boldsymbol{x}) \geq \mathbb{E}_{q(\boldsymbol{z}|\boldsymbol{x})}[\log p(\boldsymbol{x}|\boldsymbol{z})] - D_{KL}(q(\boldsymbol{z}|\boldsymbol{x}) | p(\boldsymbol{z}))$$
The first term is the reconstruction objective, and the second is a KL-divergence regularizer encouraging diverse, disentangled latent codes. Key molecular VAEs include ChemVAE (SMILES-based), JT-VAE (junction tree graphs), and GrammarVAE (grammar-constrained SMILES).
Normalizing Flows (NFs)
NFs model $p(\boldsymbol{x})$ via an invertible, deterministic mapping between data and latent space, using the change-of-variable formula with Jacobian determinants. Molecular applications include GraphNVP, MoFlow (one-shot graph generation), GraphAF (autoregressive flow), and GraphDF (discrete flow).
Generative Adversarial Networks (GANs)
GANs use a generator-discriminator game where the generator produces molecules and the discriminator distinguishes real from generated samples. Molecular GANs include MolGAN (graph-based with RL reward), ORGAN (SMILES-based with RL), and Mol-CycleGAN (molecule-to-molecule translation).
Diffusion Models
Diffusion models learn to reverse a gradual noising process. The forward process adds Gaussian noise over $T$ steps; a neural network learns to denoise at each step. The training objective reduces to predicting the noise added at each step:
$$\mathcal{L}_t = \mathbb{E}_{\boldsymbol{x}_0, \boldsymbol{\epsilon}}\left[|\epsilon_t - \epsilon_\theta(\sqrt{\bar{\alpha}_t}\boldsymbol{x}_0 + \sqrt{1 - \bar{\alpha}_t}\epsilon_t, t)|^2\right]$$
Diffusion has been particularly successful for 3D conformation generation (ConfGF, GeoDiff, DGSM).
Energy-Based Models (EBMs)
EBMs define $p(\boldsymbol{x}) = \frac{\exp(-E_\theta(\boldsymbol{x}))}{A}$ where $E_\theta$ is a learned energy function. The challenge is computing the intractable partition function $A$, addressed via contrastive divergence, noise-contrastive estimation, or score matching.
Combinatorial Optimization Methods
Unlike DGMs that learn from data distributions, combinatorial optimization methods (COMs) search directly over discrete chemical space using oracle calls to evaluate candidate molecules.
Reinforcement Learning (RL)
RL formulates molecule generation as a Markov Decision Process: states are partial molecules, actions are adding/removing atoms or bonds, and rewards come from property oracles. Methods include GCPN (graph convolutional policy network), MolDQN (deep Q-network), RationaleRL (property-aware substructure assembly), and REINVENT (SMILES-based policy gradient).
Genetic Algorithms (GA)
GAs maintain a population of molecules and evolve them through mutation and crossover operations. GB-GA operates on molecular graphs, GA+D uses SELFIES with adversarial discriminator enhancement, and JANUS uses SELFIES with parallel exploration strategies.
Bayesian Optimization (BO)
BO builds a Gaussian process surrogate of the objective function and uses an acquisition function to decide which molecules to evaluate next. It is often combined with VAE latent spaces (Constrained-BO-VAE, MSO) to enable continuous optimization.
Monte Carlo Tree Search (MCTS)
MCTS explores the molecular construction tree by branching and evaluating promising intermediates. ChemTS and MP-MCTS combine MCTS with autoregressive SMILES generators.
MCMC Sampling
MCMC methods (MIMOSA, MARS) formulate molecule optimization as sampling from a target distribution defined by multiple property objectives, using graph neural networks as proposal distributions.
Other Approaches
The survey also identifies two additional paradigms that do not fit neatly into either DGM or COM categories. Optimal Transport (OT) is used when matching between groups of molecules, particularly for conformation generation where each molecule has multiple associated 3D structures (e.g., GeoMol, EquiBind). Differentiable Learning formulates discrete molecules as differentiable objects, enabling gradient-based continuous optimization directly on molecular graphs (e.g., DST).
Task Taxonomy: Eight Molecule Generation Tasks
The survey’s central organizational contribution is a unified taxonomy of eight distinct molecule design tasks, defined by three axes: (1) whether generation is de novo (from scratch, no reference molecule) or conditioned on an input molecule, (2) whether the goal is generation (distribution learning, producing valid and diverse molecules) or optimization (goal-directed search for molecules with specific properties), and (3) the input/output data representation (1D string, 2D graph, 3D geometry). The paper’s Table 2 maps all combinations of these axes, showing that many are not meaningful (e.g., 1D string input to 2D graph output with no goal). Only eight combinations correspond to active research areas.
1D/2D Tasks
- De novo 1D/2D molecule generation: Generate new molecules from scratch to match a training distribution. Methods span VAEs (ChemVAE, JT-VAE), flows (GraphNVP, MoFlow, GraphAF), GANs (MolGAN, ORGAN), ARs (MolecularRNN), and EBMs (GraphEBM).
- De novo 1D/2D molecule optimization: Generate molecules with optimal properties from scratch, using oracle feedback. Methods include RL (GCPN, MolDQN), GA (GB-GA, JANUS), MCTS (ChemTS), and MCMC (MIMOSA, MARS).
- 1D/2D molecule optimization: Optimize properties of a given input molecule via local search. Methods include graph-to-graph translation (VJTNN, CORE, MOLER), VAE+BO (MSO, Constrained-BO-VAE), GANs (Mol-CycleGAN, LatentGAN), and differentiable approaches (DST).
3D Tasks
- De novo 3D molecule generation: Generate novel 3D molecular structures from scratch, respecting geometric validity. Methods include ARs (G-SchNet, G-SphereNet), VAEs (3DMolNet), flows (E-NFs), and RL (MolGym).
- De novo 3D conformation generation: Generate 3D conformations from given 2D molecular graphs. Methods include VAEs (CVGAE, ConfVAE), diffusion models (ConfGF, GeoDiff, DGSM), and optimal transport (GeoMol).
- De novo binding-based 3D molecule generation: Design 3D molecules for specific protein binding pockets. Methods include density-based VAEs (liGAN), RL (DeepLigBuilder), and ARs (3DSBDD).
- De novo binding-pose conformation generation: Find the appropriate 3D conformation of a given molecule for a given protein pocket. Methods include EBMs (DeepDock) and optimal transport (EquiBind).
- 3D molecule optimization: Optimize 3D molecular properties (scaffold replacement, conformation refinement). Methods include BO (BOA), ARs (3D-Scaffold, cG-SchNet), and VAEs (Coarse-GrainingVAE).
Evaluation Metrics
The survey organizes evaluation metrics into four categories.
Generation Evaluation
Basic metrics assess the quality of generated molecules:
- Validity: fraction of chemically valid molecules among all generated molecules
- Novelty: fraction of generated molecules absent from the training set
- Uniqueness: fraction of distinct molecules among generated samples
- Quality: fraction passing a predefined chemical rule filter
- Diversity (internal/external): measured via pairwise similarity (Tanimoto, scaffold, or fragment) within generated set and between generated and training sets
Distribution Evaluation
Metrics measuring how well generated molecules capture the training distribution: KL divergence over physicochemical descriptors, Frechet ChemNet Distance (FCD), and Mean Maximum Discrepancy (MMD).
Optimization Evaluation
Property oracles used as optimization targets: Synthetic Accessibility (SA), Quantitative Estimate of Drug-likeness (QED), LogP, kinase inhibition scores (GSK3-beta, JNK3), DRD2 activity, GuacaMol benchmark oracles, and Vina docking scores. Constrained optimization additionally considers structural similarity to reference molecules via Tanimoto, scaffold, or fragment similarity.
3D Evaluation
3D-specific metrics include stability (matching valence rules in 3D), RMSD and Kabsch-RMSD (conformation alignment), and Coverage/Matching scores for conformation ensembles.
Datasets
The survey catalogs 12 major datasets spanning 1D/2D and 3D molecule generation:
| Dataset | Scale | Dimensionality | Purpose |
|---|---|---|---|
| ZINC | 250K | 1D/2D | Virtual screening compounds |
| ChEMBL | 2.1M | 1D/2D | Bioactive molecules |
| MOSES | 1.9M | 1D/2D | Benchmarking generation |
| CEPDB | 4.3M | 1D/2D | Organic photovoltaics |
| GDB13 | 970M | 1D/2D | Enumerated small molecules |
| QM9 | 134K | 1D/2D/3D | Quantum chemistry properties |
| GEOM | 450K/37M | 1D/2D/3D | Conformer ensembles |
| ISO17 | 200/431K | 1D/2D/3D | Molecule-conformation pairs |
| Molecule3D | 3.9M | 1D/2D/3D | DFT ground-state geometries |
| CrossDock2020 | 22.5M | 1D/2D/3D | Docked ligand poses |
| scPDB | 16K | 1D/2D/3D | Binding sites |
| DUD-E | 23K | 1D/2D/3D | Active compounds with decoys |
Challenges and Opportunities
Challenges
- Out-of-distribution generation: Most deep generative models imitate known molecule distributions and struggle to explore truly novel chemical space.
- Unrealistic problem formulation: Many task setups do not respect real-world chemistry constraints.
- Expensive oracle calls: Methods typically assume unlimited access to property evaluators, which is unrealistic in drug discovery.
- Lack of interpretability: Few methods explain why generated molecules have desired properties. Quantitative interpretability evaluation remains an open problem.
- No unified evaluation protocols: The field lacks consensus on what defines a “good” drug candidate and how to fairly compare methods.
- Insufficient benchmarking: Despite the enormous chemical space ($10^{23}$ to $10^{60}$ drug-like molecules), available benchmarks use only small fractions of large databases.
- Low-data regime: Many real-world applications have limited training data, and generating molecules under data scarcity remains difficult.
Opportunities
- Extension to complex structured data: Techniques from small molecule generation may transfer to proteins, antibodies, genes, crystal structures, and polysaccharides.
- Connection to later drug development phases: Bridging the gap between molecule design and preclinical/clinical trial outcomes could improve real-world impact.
- Knowledge discovery: Generative models over molecular latent spaces could reveal chemical rules governing molecular properties, and graph structure learning could uncover implicit non-bonded interactions.
Limitations
- The survey was published in March 2022, so it does not cover subsequent advances in diffusion models for molecules (e.g., EDM, DiffSBDD), large language models applied to chemistry, or flow matching approaches.
- Coverage focuses on small molecules. Macromolecule design (proteins, nucleic acids) is noted as a future direction rather than surveyed.
- The survey catalogs methods but does not provide head-to-head experimental comparisons across all 100+ methods. Empirical discussion relies on individual papers’ reported results.
- 1D string-based methods receive less detailed coverage than graph and geometry-based approaches, reflecting the field’s shift toward structured representations at the time of writing.
- As a survey, this paper produces no code, models, or datasets. The surveyed methods’ individual repositories are referenced in their original publications but are not aggregated here.