Paper Information
Citation: Fang, Y., Zhang, N., Chen, Z., Guo, L., Fan, X., & Chen, H. (2024). Domain-Agnostic Molecular Generation with Chemical Feedback. Proceedings of the Twelfth International Conference on Learning Representations (ICLR 2024).
Publication: ICLR 2024
Additional Resources:
A SELFIES-Based Method for Molecular Generation
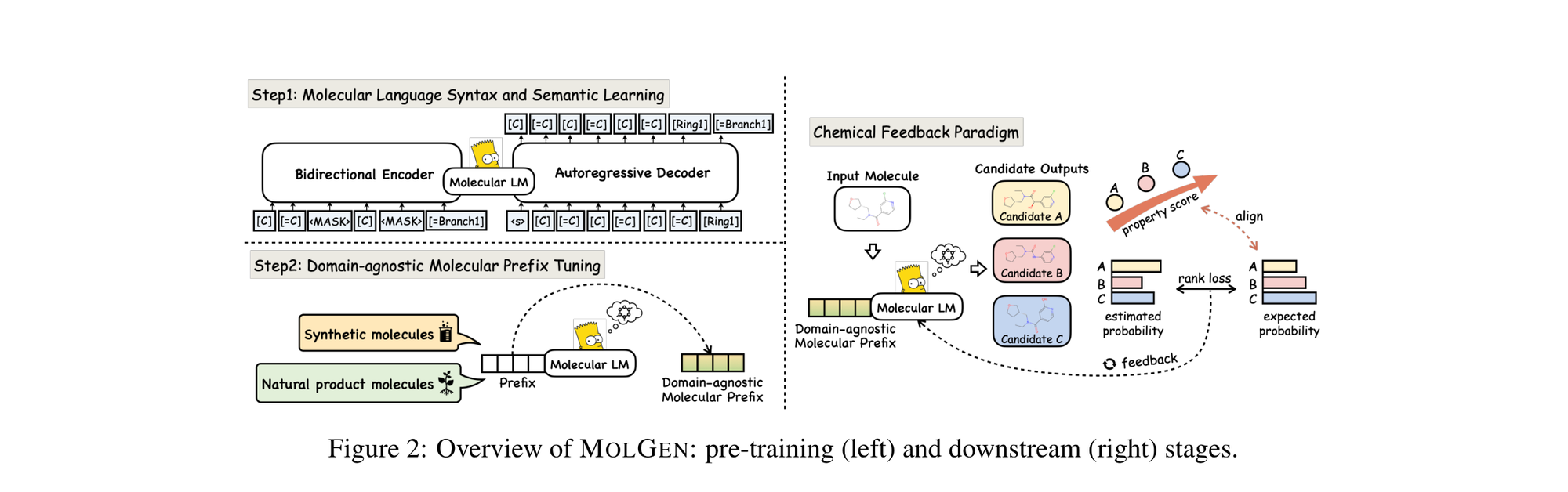
This is a Method paper that introduces MolGen, a pre-trained molecular language model for generating molecules with desired chemical properties. The primary contribution is a three-part framework: (1) pre-training on 100M+ molecular SELFIES to learn structural and grammatical knowledge, (2) domain-agnostic molecular prefix tuning for cross-domain knowledge transfer, and (3) a chemical feedback paradigm that aligns the model’s generative probabilities with real-world chemical preferences. MolGen is the first language model pre-trained on SELFIES rather than SMILES, which guarantees 100% syntactic validity of generated molecules.
Challenges in Language Model-Based Molecule Generation
Generating novel molecules with desirable properties is a central task in drug discovery and chemical design. The molecular space is estimated at $10^{33}$ possible structures, making exhaustive search impractical. Prior deep generative approaches face several limitations:
- Syntactic invalidity: SMILES-based language models frequently generate strings that do not correspond to valid molecular graphs. A single random mutation of a SMILES string has only a 9.9% chance of remaining valid.
- Narrow domain focus: Most existing models focus exclusively on synthetic molecules and neglect natural products, which have distinct structural complexity and scaffold diversity.
- Molecular hallucinations: Generated molecules may satisfy chemical structural rules yet fail to exhibit anticipated chemical activity in practical applications. The authors formally define this as molecules that “comply with chemical structural rules, yet fail to exhibit practical utility or the anticipated properties.”
- Limited optimization signals: Existing approaches rely on reinforcement learning (high variance), fixed-dimensional latent spaces, or expert-provided generation rules, all of which impede efficient exploration of chemical space.
Core Innovation: Pre-training with SELFIES and Chemical Feedback
MolGen’s novelty rests on three interconnected components.
SELFIES-Based Pre-training
MolGen uses SELFIES (Self-Referencing Embedded Strings) instead of SMILES. SELFIES guarantees that every possible combination of symbols in the alphabet corresponds to a chemically valid molecular graph. The model uses a compact vocabulary of 185 tokens.
The first pre-training stage uses a BART-style encoder-decoder. Tokens from a SELFIES string $S = {s_1, \ldots, s_l}$ are randomly replaced with [MASK], then the corrupted input is encoded bidirectionally and decoded left-to-right. The reconstruction loss is:
$$ \mathcal{L}_{\text{ce}}(S) = -\sum_{j=1}^{l} \sum_{s} p_{\text{true}}(s \mid S, S_{< j}) \log p_{\theta}(s \mid S, S_{< j}; \theta) $$
where $S_{< j}$ denotes the partial sequence ${s_0, \ldots, s_{j-1}}$ and $p_{\text{true}}$ is the one-hot distribution under standard maximum likelihood estimation.
Domain-Agnostic Molecular Prefix Tuning
The second pre-training stage introduces shared prefix vectors $P_k, P_v \in \mathbb{R}^{m \times d}$ prepended to the keys and values of multi-head attention at each layer. Unlike conventional prefix tuning that freezes model parameters, MolGen updates the entire model. The attention output becomes:
$$ \text{head} = \text{Attn}\left(xW_q, [P_k, XW_k], [P_v, XW_v]\right) $$
This decomposes into a linear interpolation between prefix attention and standard attention:
$$ \text{head} = \lambda(x) \cdot \text{Attn}(xW_q, P_k, P_v) + (1 - \lambda(x)) \cdot \text{Attn}(xW_q, XW_k, XW_v) $$
where $\lambda(x)$ is a scalar representing the sum of normalized attention weights on the prefixes. The prefixes are trained simultaneously across synthetic and natural product domains, acting as a domain instructor.
Chemical Feedback Paradigm
To address molecular hallucinations, MolGen aligns the model’s probabilistic rankings with chemical preference rankings. Given a molecule $S$ and a set of candidate outputs $\mathcal{S}^*$ with distinct property scores $\text{Ps}(\cdot)$, the model should satisfy:
$$ p_{\text{true}}(S_i \mid S) > p_{\text{true}}(S_j \mid S), \quad \forall S_i, S_j \in \mathcal{S}^*, \text{Ps}(S_i) > \text{Ps}(S_j) $$
This is enforced via a rank loss:
$$ \mathcal{L}_{\text{rank}}(S) = \sum_{i} \sum_{j > i} \max\left(0, f(S_j) - f(S_i) + \gamma_{ij}\right) $$
where $\gamma_{ij} = (j - i) \cdot \gamma$ is a margin scaled by rank difference and $f(S) = \sum_{t=1}^{l} \log p_{\theta}(s_t \mid S, S_{< t}; \theta)$ is the estimated log-probability. The overall training objective combines cross-entropy and rank loss:
$$ \mathcal{L} = \mathcal{L}_{\text{ce}} + \alpha \mathcal{L}_{\text{rank}} $$
Label smoothing is applied to the target distribution in $\mathcal{L}_{\text{ce}}$, allocating probability mass $\beta$ to non-target tokens to maintain generative diversity.
Experiments Across Distribution Learning and Property Optimization
Datasets
- Stage 1 pre-training: 100M+ unlabeled molecules from ZINC-15 (molecular weight $\leq$ 500 Da, LogP $\leq$ 5)
- Stage 2 pre-training: 2.22M molecules spanning synthetic (ZINC, MOSES) and natural product (NPASS, 30,926 compounds) domains
- Downstream evaluation: MOSES synthetic dataset, ZINC250K, and natural product molecules
Molecular Distribution Learning
MolGen generates 10,000 synthetic and 80,000 natural product molecules, evaluated on seven metrics (Validity, Fragment similarity, Scaffold similarity, SNN, Internal Diversity, FCD, and Novelty). Baselines include AAE, LatentGAN, CharRNN, VAE, JT-VAE, LIMO, and Chemformer.
| Model | Validity | Frag | Scaf | SNN | IntDiv | FCD | Novelty |
|---|---|---|---|---|---|---|---|
| Chemformer | .9843 | .9889 | .9248 | .5622 | .8553 | .0061 | .9581 |
| MolGen | 1.000 | .9999 | .9999 | .9996 | .8567 | .0015 | 1.000 |
On synthetic molecules, MolGen achieves 100% validity, near-perfect fragment and scaffold similarity, and the lowest FCD (0.0015). For natural products, MolGen achieves FCD of 0.6519 compared to Chemformer’s 0.8346.
Targeted Molecule Discovery
For penalized logP maximization (top-3 scores):
| Model | 1st | 2nd | 3rd |
|---|---|---|---|
| MARS (no length limit) | 44.99 | 44.32 | 43.81 |
| MolGen (no length limit) | 80.30 | 74.70 | 69.85 |
| MolGen (length-limited) | 30.51 | 28.98 | 28.95 |
For QED maximization, MolGen achieves the maximum score of 0.948 across the top-3.
Molecular Docking
MolGen optimizes binding affinity for two protein targets (ESR1 and ACAA1), measured by dissociation constant $K_D$ (lower is better):
| Model | ESR1 1st | ESR1 2nd | ESR1 3rd | ACAA1 1st | ACAA1 2nd | ACAA1 3rd |
|---|---|---|---|---|---|---|
| LIMO | 0.72 | 0.89 | 1.4 | 37 | 37 | 41 |
| MolGen | 0.13 | 0.35 | 0.47 | 3.36 | 3.98 | 8.50 |
MolGen achieves the lowest dissociation constants across both targets. Optimization of the 1,000 worst-affinity molecules yields 96.7% relative improvement for ESR1 and 70.4% for ACAA1.
Constrained Molecular Optimization
Optimizing 800 molecules from ZINC250K with lowest p-logP scores under Tanimoto similarity constraints:
| Model | $\delta = 0.6$ | $\delta = 0.4$ |
|---|---|---|
| RetMol | 3.78 (3.29) | 11.55 (11.27) |
| MolGen | 12.08 (0.82) | 12.35 (1.21) |
MolGen achieves the highest mean improvement with the lowest standard deviation under both constraints.
Ablation Studies
- Chemical feedback: Without it, the model generates molecules with property scores similar to initial molecules. With it ($\alpha = 3$), property scores increase progressively across generation rounds.
- Prefix tuning: Removing prefix tuning reduces constrained optimization improvement by 0.45 at $\delta = 0.6$ and 2.12 at $\delta = 0.4$.
- Label smoothing: Enhances diversity of generated molecules as measured by Internal Diversity.
- Substructure attention: MolGen focuses attention on chemically meaningful functional groups (fluoro, phenyl, hydroxyl), while SMILES-based PLMs scatter attention across syntactic tokens. The Substructure Attention Level (SAL) metric confirms MolGen’s superior focus.
Key Findings, Limitations, and Future Directions
Key Findings
- SELFIES pre-training guarantees 100% molecular validity, eliminating the need for external valency checks.
- Domain-agnostic prefix tuning enables effective knowledge transfer between synthetic and natural product domains.
- The chemical feedback paradigm aligns model outputs with chemical preferences without requiring external annotated data or reference databases.
- MolGen achieves the best or competitive results across all evaluated tasks: distribution learning, targeted molecule discovery, constrained optimization, and molecular docking.
Limitations
The authors acknowledge several limitations:
- Computational cost: Training and fine-tuning on large datasets is computationally intensive.
- Model interpretability: The transformer architecture makes it difficult to understand explicit rationale behind decisions.
- Single-target optimization only: The chemical feedback paradigm handles single-target optimization; multiple conflicting objectives could create ambiguous optimization trajectories.
- Task specificity: MolGen is designed for 2D molecular generation; 3D conformation information is not incorporated.
- Reaction prediction: When applied to reaction prediction (an off-target task), MolGen achieves only 71.4% accuracy on 39,990 reaction samples.
Future Directions
The authors suggest applying MolGen to retrosynthesis and reaction prediction, exploring multimodal pre-training, and incorporating additional knowledge sources.
Reproducibility Details
Data
| Purpose | Dataset | Size | Notes |
|---|---|---|---|
| Stage 1 pre-training | ZINC-15 | 100M+ molecules | MW $\leq$ 500 Da, LogP $\leq$ 5 |
| Stage 2 pre-training | ZINC + MOSES + NPASS | 2.22M molecules | Synthetic and natural product domains |
| Distribution learning (synthetic) | MOSES | ~1.9M molecules | Standard benchmark split |
| Distribution learning (natural) | NPASS | 30,926 compounds | 30,126 train / 800 test |
| Constrained optimization | ZINC250K | 800 molecules | Lowest p-logP scores |
Algorithms
- Architecture: BART-based encoder-decoder with SELFIES vocabulary (185 tokens)
- Prefix length: 5 tunable vectors per layer
- Optimizer: LAMB (pre-training), AdamW (fine-tuning)
- Pre-training: 600M steps with linear warm-up (180,000 steps) followed by linear decay
- Rank loss weight ($\alpha$): Recommended values of 3 or 5
- Candidate generation: 30 candidates per molecule (synthetic), 8 candidates (natural products)
Models
MolGen is publicly available on Hugging Face. The model uses a vocabulary of 185 SELFIES tokens and is comparable in size to Chemformer-large.
Evaluation
| Metric | Domain | MolGen | Best Baseline | Notes |
|---|---|---|---|---|
| FCD (lower is better) | Synthetic | 0.0015 | 0.0061 (Chemformer) | Distribution learning |
| p-logP top-1 (no limit) | Synthetic | 80.30 | 44.99 (MARS) | Targeted discovery |
| QED top-1 | Synthetic | 0.948 | 0.948 (several) | Tied at maximum |
| ESR1 $K_D$ top-1 | Docking | 0.13 | 0.72 (LIMO) | Binding affinity |
| p-logP improvement ($\delta=0.4$) | Synthetic | 12.35 (1.21) | 11.55 (11.27) (RetMol) | Constrained optimization |
Hardware
- 6 NVIDIA V100 GPUs
- Pre-training batch size: 256 molecules per GPU
- Fine-tuning batch size: 6 (synthetic and natural product)
- Training: 100 epochs for fine-tuning tasks
Artifacts
| Artifact | Type | License | Notes |
|---|---|---|---|
| zjunlp/MolGen | Code | Unknown | Official PyTorch implementation |
| zjunlp/MolGen-large | Model | Unknown | Pre-trained weights on Hugging Face |
Citation
@inproceedings{fang2024domain,
title={Domain-Agnostic Molecular Generation with Chemical Feedback},
author={Fang, Yin and Zhang, Ningyu and Chen, Zhuo and Guo, Lingbing and Fan, Xiaohui and Chen, Huajun},
booktitle={The Twelfth International Conference on Learning Representations},
year={2024},
url={https://openreview.net/forum?id=9rPyHyjfwP}
}