Paper Information
Citation: Ross, J., Belgodere, B., Chenthamarakshan, V., Padhi, I., Mroueh, Y., & Das, P. (2022). Large-Scale Chemical Language Representations Capture Molecular Structure and Properties. Nature Machine Intelligence, 4, 1256-1264. https://doi.org/10.1038/s42256-022-00580-7
Publication: Nature Machine Intelligence 2022
Additional Resources:
A Billion-Scale Chemical Language Model
This is primarily a Method paper ($\Psi_{\text{Method}}$).
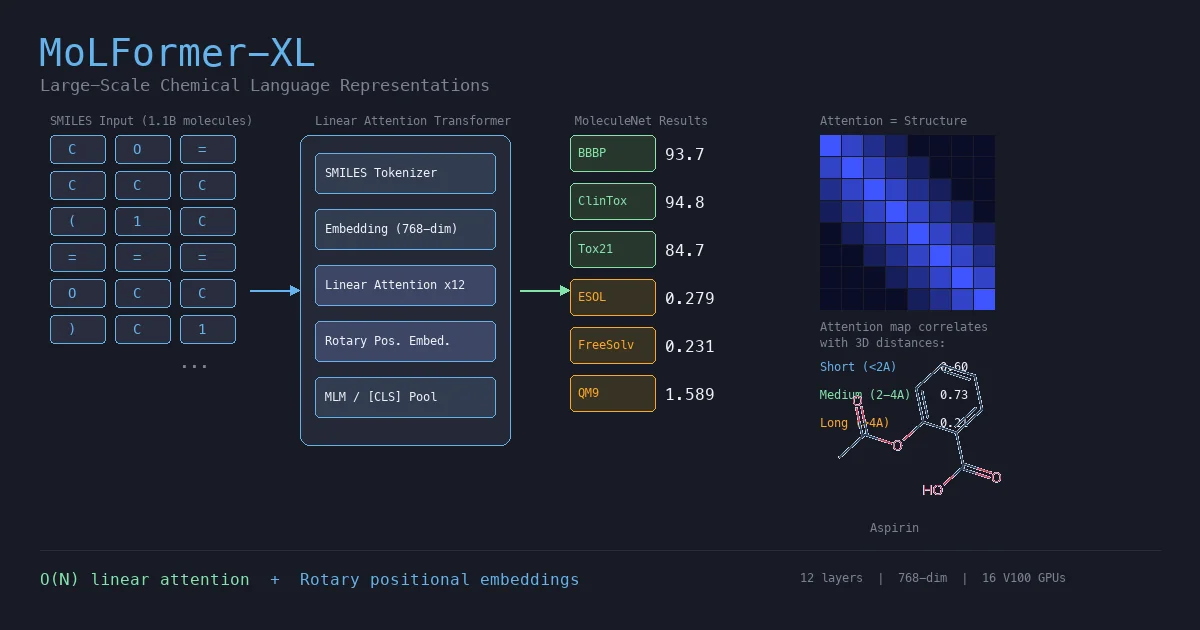
MoLFormer is a transformer encoder pretrained via masked language modeling on 1.1 billion SMILES strings from PubChem and ZINC. The key architectural choices are linear attention (for $O(N)$ complexity instead of $O(N^2)$) and rotary positional embeddings (RoPE). The resulting model, MoLFormer-XL, produces molecular embeddings that outperform or match GNN baselines across a wide range of MoleculeNet classification and regression tasks, including quantum-chemical property prediction from SMILES alone.
Bridging the Gap Between Molecular Languages and Graph Neural Networks
Prior chemical language models like ChemBERTa were pretrained on relatively small datasets (10M-77M molecules) and generally underperformed GNNs on molecular property prediction. The core question: does a transformer trained on a sufficiently large SMILES corpus learn enough chemical structure to compete with graph-based methods that have explicit topological inductive biases?
Two specific challenges motivated this work:
- Scale: The chemical space spans $10^{60}$ to $10^{100}$ plausible molecules, yet labeled property data is scarce. Self-supervised pretraining on the ~1.1B unlabeled molecules available in public databases could provide a general-purpose representation.
- Efficiency: Standard transformer attention is $O(N^2)$ in sequence length, making billion-scale pretraining impractical without architectural modifications.
Linear Attention with Rotary Positional Embeddings
MoLFormer’s two key architectural choices are its attention mechanism and positional encoding scheme.
Standard attention computes:
$$ \text{Attention}_m(Q, K, V) = \frac{\sum_{n=1}^{N} \exp(\langle q_m, k_n \rangle) v_n}{\sum_{n=1}^{N} \exp(\langle q_m, k_n \rangle)} $$
MoLFormer replaces this with linear attention using a generalized feature map $\varphi$, combined with rotary positional embeddings $R_m$ applied before the feature map:
$$ \text{Attention}_m(Q, K, V) = \frac{\sum_{n=1}^{N} \langle \varphi(R_m q_m), \varphi(R_n k_n) \rangle v_n}{\sum_{n=1}^{N} \langle \varphi(R_m q_m), \varphi(R_n k_n) \rangle} $$
This differs from the original RoFormer formulation, which applies the rotation after the feature map. The authors found that rotating the raw queries and keys before projection led to faster convergence and lower validation loss. The combination of linear attention and adaptive sequence-length bucketing reduces GPU requirements from ~1000 to 16 for training on the full 1.1B corpus.
The model uses masked language modeling (15% token masking, following BERT conventions) with a vocabulary of 2,362 SMILES tokens. Sequence length is capped at 202 tokens, covering 99.4% of all molecules.
Broad MoleculeNet Benchmarking with Scaling Ablations
MoLFormer-XL was evaluated on 11 MoleculeNet tasks against supervised GNNs, self-supervised GNNs, and prior language models.
Classification tasks (ROC-AUC, scaffold split; values reported as percentages in the original paper, converted to proportions here for consistency):
| Model | BBBP | Tox21 | ClinTox | HIV | BACE | SIDER |
|---|---|---|---|---|---|---|
| MoLFormer-XL | 0.937 | 0.847 | 0.948 | 0.822 | 0.882 | 0.690 |
| N-Gram | 0.912 | 0.769 | 0.855 | 0.830 | 0.876 | 0.632 |
| MolCLR | 0.736 | 0.798 | 0.932 | 0.806 | 0.890 | 0.680 |
| GEM | 0.724 | 0.781 | 0.901 | 0.806 | 0.856 | 0.672 |
| Hu et al. | 0.708 | 0.787 | 0.789 | 0.802 | 0.859 | 0.652 |
| GeomGCL | - | 0.850 | 0.919 | - | - | 0.648 |
| ChemBERTa | 0.643 | - | 0.906 | 0.622 | - | - |
Regression tasks (RMSE for ESOL/FreeSolv/Lipophilicity, avg MAE for QM9/QM8):
| Model | QM9 | QM8 | ESOL | FreeSolv | Lipophilicity |
|---|---|---|---|---|---|
| MoLFormer-XL | 1.5894 | 0.0102 | 0.2787 | 0.2308 | 0.5289 |
| A-FP | 2.6355 | 0.0282 | 0.5030 | 0.736 | 0.578 |
| MPNN | 3.1898 | 0.0143 | 0.58 | 1.150 | 0.7190 |
| GC | 4.3536 | 0.0148 | 0.970 | 1.40 | 0.655 |
MoLFormer-XL also outperforms geometry-aware GNNs (DimeNet, GeomGCL, GEM) on ESOL (0.279 vs 0.575), FreeSolv (0.231 vs 0.866), and Lipophilicity (0.529 vs 0.541).
Key ablation findings:
- Data scale matters: Performance improves monotonically from 10% subsets through the full 1.1B corpus. Training on 100% ZINC alone performed worst, likely due to its smaller vocabulary and less diverse molecule lengths.
- Model depth matters: MoLFormer-Base (6 layers) underperforms MoLFormer-XL (12 layers) on most tasks.
- Fine-tuning » frozen: Fine-tuning the full encoder consistently outperforms using frozen embeddings with a downstream classifier.
- Rotary > absolute at scale: Rotary embeddings underperform absolute embeddings on smaller pretraining sets but overtake them once the corpus exceeds 1B molecules.
SMILES Transformers Learn Molecular Geometry
The most striking finding is that MoLFormer’s attention patterns correlate with 3D interatomic distances, despite training only on 1D SMILES strings.
Using QM9 molecules with known 3D geometries, the authors computed cosine similarity between attention maps and spatial distance matrices across three distance categories:
| Distance Category | Range | Linear Attention (Rotary) | Full Attention (Rotary) |
|---|---|---|---|
| Short | $\leq$ 2 Å | 0.594-0.602 | 0.598-0.615 |
| Medium | 2-4 Å | 0.724-0.730 | 0.716-0.727 |
| Long | 4-10 Å | 0.209-0.211 | 0.204-0.210 |
The strong correlation in the short and medium categories indicates the model captures covalent bond connectivity and near-neighbor spatial relationships. Linear attention shows marginally higher cosine similarity than full attention on medium-range distances (0.724-0.730 vs 0.716-0.727), though the differences are small.
MoLFormer-XL embeddings also correlate more strongly with molecular fingerprint similarity (0.64 vs 0.48 for ChemBERTa) and maximum common subgraph size (-0.60 vs -0.44), confirming that the representations encode structural information.
Limitations:
- Quantum-chemical energies: SchNet and DimeNet (which encode explicit 3D geometry) outperform MoLFormer-XL on QM9 atomization energy tasks, with DimeNet achieving roughly 10x lower MAE on U0_atom (0.008 vs 0.083 eV). 3D information remains important for these properties.
- Sequence length cap: The 202-token limit excludes 0.6% of molecules, potentially limiting applicability to larger structures.
- SMILES canonicalization: The model depends on RDKit canonical SMILES; sensitivity to non-canonical forms is not evaluated.
Reproducibility Details
Data
| Purpose | Dataset | Size | Notes |
|---|---|---|---|
| Pretraining | PubChem | 111M molecules | Canonical SMILES via RDKit |
| Pretraining | ZINC | ~1B molecules | Canonical SMILES via RDKit |
| Pretraining (combined) | PubChem + ZINC | ~1.1B molecules | MoLFormer-XL training set |
| Classification | BBBP, Tox21, ClinTox, HIV, BACE, SIDER | 1,427-41,127 | MoleculeNet scaffold splits |
| Regression | QM9, QM8, ESOL, FreeSolv, Lipophilicity | 642-133,885 | MoleculeNet random splits (QM9/QM8), scaffold (others) |
Algorithms
- Pretraining objective: Masked language modeling (15% selection: 80% masked, 10% random, 10% unchanged)
- Tokenization: SMILES tokenizer from Schwaller et al., vocabulary of 2,362 tokens
- Sequence length: 1-202 tokens (99.4% coverage)
- Optimizer: Fused LAMB (via APEX), chosen for stability with large batch sizes and no need for learning rate warm-up
- Adaptive bucketing: Sequences grouped by length into buckets to minimize padding waste
Models
- Architecture: Transformer encoder with linear attention and rotary positional embeddings
- MoLFormer-XL: 12 layers, 12 attention heads, hidden size 768
- MoLFormer-Base: 6 layers (ablation only)
- Feature map size: 32 (generalized feature map for linear attention)
- Frozen head: Fully connected model with hyperparameter sweep (learning rate, batch size, hidden dim, number of layers)
Evaluation
| Metric | Task Type | Details |
|---|---|---|
| ROC-AUC | Classification | Scaffold splits per MoleculeNet |
| RMSE | Regression (ESOL, FreeSolv, Lipophilicity) | Scaffold splits |
| Avg MAE | Regression (QM9, QM8) | Random splits per MoleculeNet |
QM9 results also reported with 5-fold cross-validation for robustness.
Hardware
- Compute: GPU cluster with nodes containing either 8 NVIDIA Tesla V100 (32GB) or 8 Ampere A100 (40GB) GPUs connected via NVLink and InfiniBand
- GPU reduction: Linear attention + bucketing reduced GPU requirements from ~1000 to 16
Artifacts
| Artifact | Type | License | Notes |
|---|---|---|---|
| IBM/molformer | Code | Apache-2.0 | Pretraining, fine-tuning, and attention visualization |
| MoLFormer-XL (HuggingFace) | Model | Apache-2.0 | Pretrained weights (46.8M parameters) |
| PubChem | Dataset | Public domain | 111M molecules |
| ZINC | Dataset | See ZINC terms | ~1B molecules |
Citation
@article{ross2022molformer,
title={Large-Scale Chemical Language Representations Capture Molecular Structure and Properties},
author={Ross, Jerret and Belgodere, Brian and Chenthamarakshan, Vijil and Padhi, Inkit and Mroueh, Youssef and Das, Payel},
journal={Nature Machine Intelligence},
volume={4},
pages={1256--1264},
year={2022},
publisher={Nature Publishing Group},
doi={10.1038/s42256-022-00580-7}
}