Paper Contribution and Methodological Classification
This is a Method paper. It adapts the Transformer architecture to chemical reaction prediction, treating it as a machine translation problem from reactant SMILES to product SMILES. The key contributions are (1) demonstrating that a fully attention-based model outperforms all prior template-based, graph-based, and RNN-based methods, (2) showing the model works without separating reactants from reagents, and (3) introducing calibrated uncertainty estimation for ranking synthesis pathways.
Motivation: Limitations of Existing Reaction Prediction
Prior approaches to reaction prediction fell into two broad groups, template-based and template-free, each with fundamental limitations:
- Template-based methods rely on libraries of reaction rules, either handcrafted or automatically extracted from atom-mapped data. Automatic template extraction itself depends on atom mapping, which depends on templates, creating a circular dependency.
- Graph-based template-free methods (e.g., WLDN, ELECTRO) avoid explicit templates but still require atom-mapped training data and cannot handle stereochemistry.
- RNN-based seq2seq models (also template-free) treat reactions as SMILES translation but impose a positional inductive bias: tokens far apart in the SMILES string are assumed to be less related. This is incorrect because SMILES position has no relationship to 3D spatial distance.
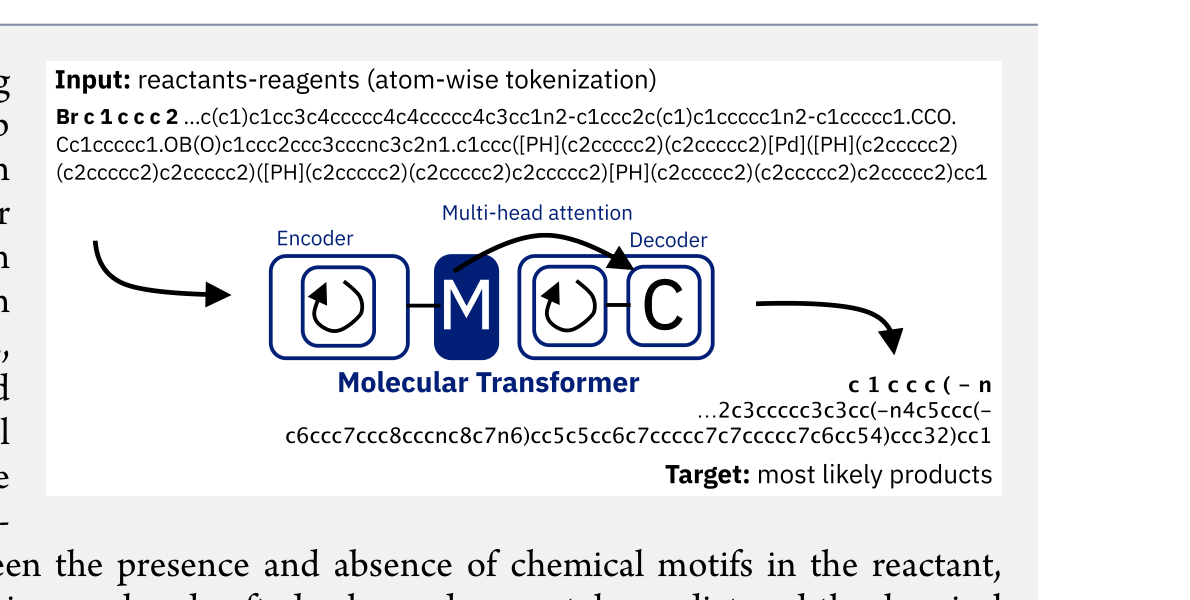
Core Innovation: Transformer for Reaction Prediction
The Molecular Transformer adapts the Transformer architecture to chemical reactions by treating SMILES strings of reactants and reagents as source sequences and product SMILES as target sequences.
- Architecture: Encoder-decoder Transformer with 4 layers, 256-dimensional hidden states, 8 attention heads, and 12M parameters (reduced from the original 65M NMT model).
- Tokenization: Atom-wise regex tokenization of SMILES strings, applied uniformly to both reactants and reagents (no special reagent tokens).
- Data augmentation: Training data is doubled by generating random (non-canonical) SMILES for each reaction, which improves top-1 accuracy by roughly 1%.
- Weight averaging: Final model weights are averaged over the last 20 checkpoints, providing a further accuracy boost without the inference cost of ensembling.
- Mixed input: Unlike all prior work that separates reactants from reagents (which implicitly assumes knowledge of the product), the Molecular Transformer operates on mixed inputs where no distinction is made.
The multihead attention mechanism is the key architectural advantage over RNNs. It allows the model to attend to any pair of tokens regardless of their position in the SMILES string, correctly capturing long-range chemical relationships that RNNs miss.
Uncertainty Estimation
A central contribution is calibrated uncertainty scoring. The product of predicted token probabilities serves as a confidence score for each prediction. This score achieves 0.89 AUC-ROC for classifying whether a prediction is correct.
An important finding: label smoothing hurts uncertainty calibration. While label smoothing (as used in the original Transformer) marginally improves top-1 accuracy (87.44% vs 87.28%), it destroys the model’s ability to distinguish correct from incorrect predictions. Setting the label smoothing parameter to 0.0 preserves calibration.
The confidence score shows no correlation with SMILES length (Pearson $r = 0.06$), confirming it is not biased against predictions of larger molecules.
Experimental Results
Forward Synthesis Prediction
| Dataset | Setting | Top-1 (%) | Top-2 (%) | Top-5 (%) |
|---|---|---|---|---|
| USPTO_MIT | separated | 90.4 | 93.7 | 95.3 |
| USPTO_MIT | mixed | 88.6 | 92.4 | 94.2 |
| USPTO_STEREO | separated | 78.1 | 84.0 | 87.1 |
| USPTO_STEREO | mixed | 76.2 | 82.4 | 85.8 |
The mixed-input model (88.6%) outperforms all prior methods that used separated inputs (best previous: WLDN5 at 85.6%).
Comparison with Quantum Chemistry
On regioselectivity of electrophilic aromatic substitution in heteroaromatics, the Molecular Transformer achieves 83% top-1 accuracy vs 81% for RegioSQM (a quantum-chemistry-based predictor), at a fraction of the computational cost.
Comparison with Human Chemists
On 80 reactions sampled across rarity bins, the Molecular Transformer achieves 87.5% top-1 accuracy vs 76.5% for the best human chemist and 72.5% for the best graph-based model (WLDN5).
Chemically Constrained Beam Search
Constraining beam search to only predict atoms present in the reactants (preventing “alchemy”) produces no change in accuracy, confirming the model has learned conservation of atoms from data alone.
Trade-offs and Limitations
- Stereochemistry: Accuracy drops significantly on USPTO_STEREO (76-78% vs 88-90% on USPTO_MIT), indicating stereochemical prediction remains challenging.
- Resolution reactions: Near-zero accuracy on resolution reactions (28.6%), where reagent information is often missing from patent data.
- Unclassified reactions: Accuracy on “unrecognized” reaction classes is 46.3%, likely reflecting noisy or mistranscribed data.
- No atom mapping: The model provides no explicit atom mapping between reactants and products, which limits interpretability for understanding reaction mechanisms.
Reproducibility Details
Data
| Purpose | Dataset | Size | Notes |
|---|---|---|---|
| Primary benchmark | USPTO_MIT | 479K | Filtered by Jin et al., no stereochemistry |
| LEF subset | USPTO_LEF | 350K | Subset of MIT with linear electron flow only |
| Stereo benchmark | USPTO_STEREO | 1.0M | Patent reactions through Sept 2016, includes stereochemistry |
| Time-split test | Pistachio_2017 | 15.4K | Non-public, reactions from 2017 |
Preprocessing: SMILES canonicalized with RDKit. Regex tokenization from Schwaller et al. (2018). Two input modes: “separated” (reactants > reagents) and “mixed” (all molecules concatenated).
Model
| Hyperparameter | Value |
|---|---|
| Layers | 4 |
| Model dimension | 256 |
| Attention heads | 8 |
| Parameters | ~12M |
| Label smoothing | 0.0 |
| Optimizer | Adam |
| Warm-up steps | 8000 |
| Batch size | ~4096 tokens |
| Beam width | 5 |
Evaluation
| Metric | Task | Key Result | Baseline |
|---|---|---|---|
| Top-1 accuracy | USPTO_MIT (sep) | 90.4% | 85.6% (WLDN5) |
| Top-1 accuracy | USPTO_MIT (mixed) | 88.6% | 80.3% (S2S RNN) |
| AUC-ROC | Uncertainty calibration | 0.89 | N/A |
| Top-1 accuracy | Regioselectivity | 83% | 81% (RegioSQM) |
| Top-1 accuracy | Human comparison | 87.5% | 76.5% (best human) |
Hardware
- Training: Single Nvidia P100 GPU, 48h for best single model
- Inference: 20 min for 40K reactions on single P100
Paper Information
Citation: Schwaller, P., Laino, T., Gaudin, T., Bolgar, P., Hunter, C. A., Bekas, C., & Lee, A. A. (2019). Molecular Transformer: A Model for Uncertainty-Calibrated Chemical Reaction Prediction. ACS Central Science, 5(9), 1572-1583. https://doi.org/10.1021/acscentsci.9b00576
Publication: ACS Central Science 2019
@article{schwallerMolecularTransformerModel2019,
title = {Molecular Transformer: A Model for Uncertainty-Calibrated Chemical Reaction Prediction},
author = {Schwaller, Philippe and Laino, Teodoro and Gaudin, Th{\'e}ophile and Bolgar, Peter and Hunter, Christopher A. and Bekas, Costas and Lee, Alpha A.},
year = 2019,
journal = {ACS Central Science},
volume = {5},
number = {9},
pages = {1572--1583},
publisher = {American Chemical Society},
doi = {10.1021/acscentsci.9b00576}
}