Protein-Targeted Drug Generation as Machine Translation
This is a Method paper that proposes using the Transformer neural network architecture for protein-specific de novo drug generation. The primary contribution is framing the problem of generating molecules that bind to a target protein as a machine translation task: translating from the “language” of amino acid sequences to the SMILES representation of candidate drug molecules. The model takes only a protein’s amino acid sequence as input and generates novel molecules with predicted binding affinity, requiring no prior knowledge of active ligands, physicochemical descriptors, or the protein’s three-dimensional structure.
Limitations of Existing Generative Drug Design Approaches
Existing deep learning methods for de novo molecule generation suffer from several limitations. Most RNN-based approaches require a library of known active compounds against the target protein to fine-tune the generator or train a reward predictor for reinforcement learning. Structure-based drug design methods require the three-dimensional structure of the target protein, which can be costly and technically difficult to obtain through protein expression, purification, and crystallization. Autoencoder-based approaches (variational and adversarial) similarly depend on prior knowledge of protein binders or their physicochemical characteristics.
The estimated drug-like molecule space is on the order of $10^{60}$, while only around $10^{8}$ compounds have been synthesized. High-throughput screening is expensive and time-consuming, and virtual screening operates only on known molecules. Computational de novo design methods often generate molecules that are hard to synthesize or restrict accessible chemical space through coded rules. A method that requires only a protein’s amino acid sequence would substantially simplify the initial stages of drug discovery, particularly for targets with limited or no information about inhibitors and 3D structure.
Sequence-to-Sequence Translation with Self-Attention
The core insight is to treat protein-targeted drug generation as a translation problem between two “languages,” applying the Transformer architecture that had demonstrated strong results in neural machine translation. The encoder maps a protein amino acid sequence $(a_1, \ldots, a_n)$ to continuous representations $\mathbf{z} = (z_1, \ldots, z_n)$, and the decoder autoregressively generates a SMILES string conditioned on $\mathbf{z}$.
The self-attention mechanism computes:
$$ \text{Attention}(Q, K, V) = \text{softmax}\left(\frac{QK^T}{\sqrt{d_k}}\right)V $$
where $d_k$ is a scaling factor. Multihead attention runs $h$ parallel attention heads:
$$ \text{head}_i = \text{Attention}(QW_i^Q, KW_i^K, VW_i^V) $$
$$ \text{Multihead}(Q, K, V) = (\text{head}_1, \ldots, \text{head}_h)W^O $$
Positional encoding uses sinusoidal functions:
$$ PE_{(pos, 2i)} = \sin\left(\frac{pos}{10000^{2i / d_{model}}}\right) $$
$$ PE_{(pos, 2i+1)} = \cos\left(\frac{pos}{10000^{2i / d_{model}}}\right) $$
The self-attention mechanism is particularly well-suited for this task for two reasons. First, protein sequences can be much longer than SMILES strings (dozens of times longer), making the ability to capture long-range dependencies essential. Second, three-dimensional structural features of the binding pocket may be formed by amino acid residues far apart in the linear sequence, and multihead attention can jointly attend to different positional aspects simultaneously.
Data, Model Architecture, and Docking Evaluation
Data
The training data was retrieved from BindingDB, filtering for interactions between proteins from Homo sapiens, Rattus norvegicus, Mus musculus, and Bos taurus with binding affinity below 100 nM (IC50, Kd, or EC50). After filtering for valid PubChem CIDs, SMILES representations, UniProt IDs, molecular weight under 1000 Da, and amino acid sequence lengths between 80 and 2050, the final dataset contained 238,147 records with 1,613 unique proteins and 154,924 unique ligand SMILES strings.
Five Monte Carlo cross-validation splits were created, with the constraint that test set proteins share less than 20% sequence similarity with training set proteins (measured via Needleman-Wunsch global alignment).
Model Configuration
The model uses the original Transformer implementation via the tensor2tensor library with:
- 4 encoder/decoder layers of size 128
- 4 attention heads
- Adam optimizer with learning rate decay from the original Transformer paper
- Batch size of 4,096 tokens
- Training for 600K epochs on a single GPU in Google Colaboratory
- Vocabulary of 71 symbols (character-level tokenization)
Beam search decoding was used with two modes: beam size 4 keeping only the top-1 result (“one per one” mode) and beam size 10 keeping all 10 results (“ten per one” mode).
Chemical Validity and Uniqueness
| Metric | One per One (avg) | Ten per One (avg) |
|---|---|---|
| Valid SMILES (%) | 90.2 | 82.6 |
| Unique SMILES (%) | 92.3 | 81.7 |
| ZINC15 match (%) | 30.6 | 17.1 |
Docking Evaluation
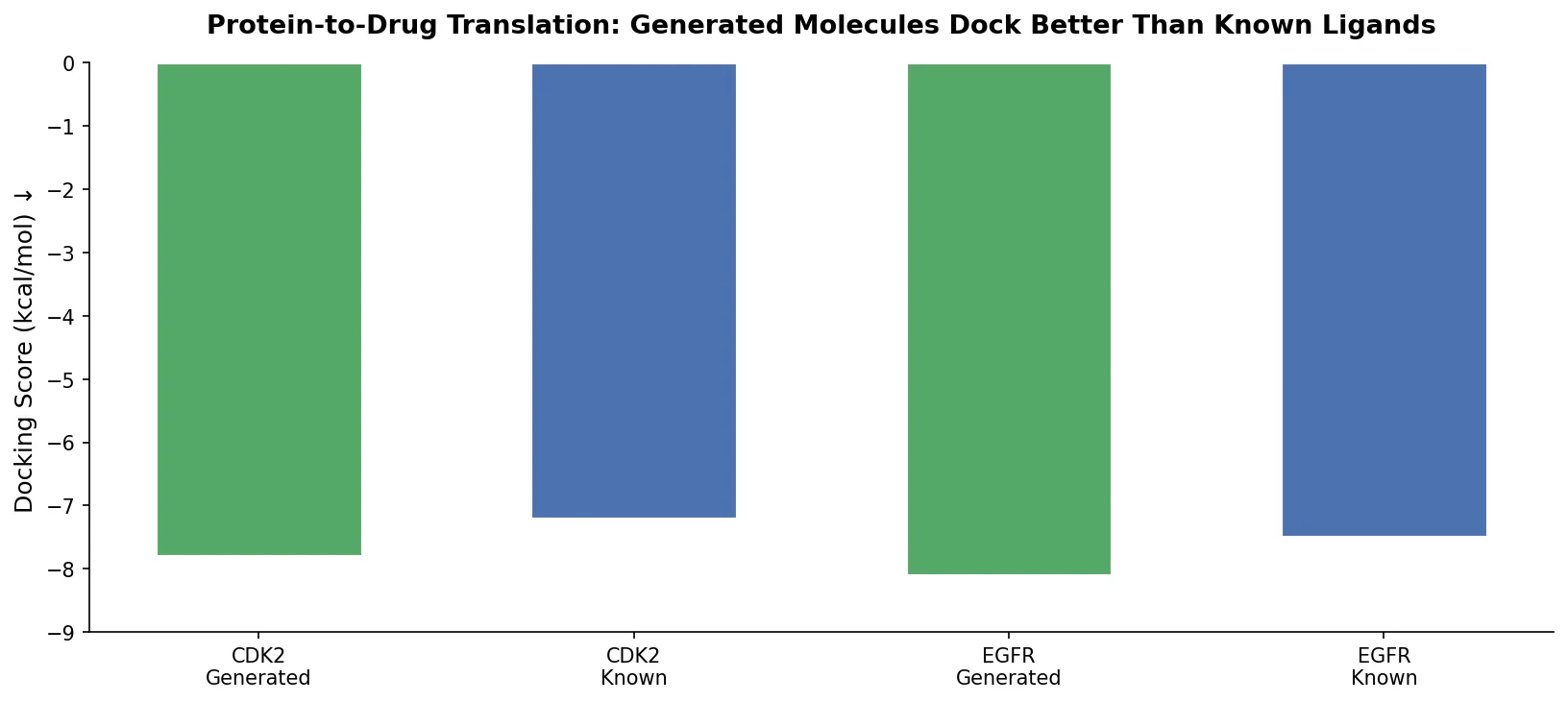
To assess binding affinity, the authors selected two receptor tyrosine kinases from the test set (IGF-1R and VEGFR2) and performed molecular docking with SMINA. Four sets of ligands were compared: known binders, randomly selected compounds, molecules generated for the target protein, and molecules generated for other targets (cross-docking control).
ROC-AUC analysis showed that the docking tool classified generated molecules for the correct target as binders at rates comparable to known binders. For the best-discriminating structures (PDB 3O23 for IGF-1R, PDB 3BE2 for VEGFR2), Mann-Whitney U tests confirmed statistically significant differences between generated-for-target molecules and random compounds, while the difference between generated-for-target and known binders was not significant (p = 0.40 and 0.26 respectively), suggesting the model generates plausible binders.
Drug-Likeness Properties
Generated molecules were evaluated against Lipinski’s Rule of Five and other drug-likeness criteria:
| Property | Constraint | One per One (%) | Ten per One (%) |
|---|---|---|---|
| logP | < 5 | 84.4 | 85.6 |
| Molecular weight | < 500 Da | 95.8 | 88.9 |
| H-bond donors | < 5 | 95.8 | 91.9 |
| H-bond acceptors | < 10 | 97.9 | 93.5 |
| Rotatable bonds | < 10 | 97.9 | 91.2 |
| TPSA | < 140 | 98.0 | 92.7 |
| SAS | < 6 | 99.9 | 100.0 |
Mean QED values were 0.66 +/- 0.19 (one per one) and 0.58 +/- 0.21 (ten per one).
Structural Novelty
Tanimoto similarity analysis showed that only 8% of generated structures had similarity above the threshold (> 0.85) to training compounds. The majority (51%) had Tanimoto scores below 0.5. The mean nearest-neighbor Tanimoto similarity of generated molecules to the training set (0.54 +/- 0.17 in one-per-one mode) was substantially lower than the mean within-training-set similarity (0.74 +/- 0.14), indicating the model generates structurally diverse molecules outside the training distribution.
Generated Molecules Show Drug-Like Properties and Predicted Binding
The model generates roughly 90% chemically valid SMILES in one-per-one mode, with 92% uniqueness. Docking simulations on IGF-1R and VEGFR2 suggest that generated molecules for the correct target are statistically indistinguishable from known binders, while molecules generated for other targets behave more like random compounds. Drug-likeness properties fall within acceptable ranges for the vast majority of generated compounds.
The authors acknowledge several limitations:
- Only two protein targets were analyzed via docking due to computational constraints, and the analysis was limited to proteins with a single well-known druggable binding pocket.
- Beam search produces molecules that differ only slightly; diverse beam search or coupling with variational/adversarial autoencoders could improve diversity.
- The fraction of molecules matching the ZINC15 database (30.6% in one-per-one mode) could potentially be reduced by pretraining on a larger compound set (e.g., ChEMBL’s 1.5 million molecules).
- Model interpretability remains limited and is identified as important future work.
- The approach is a proof of concept and requires further validation via in vitro assays across diverse protein targets.
Reproducibility Details
Data
| Purpose | Dataset | Size | Notes |
|---|---|---|---|
| Training/Test | BindingDB (filtered) | 238,147 records | 1,613 unique proteins, 154,924 unique SMILES; IC50/Kd/EC50 < 100 nM |
| Docking validation | PDB structures | 11 (IGF-1R), 20 (VEGFR2) | SMINA docking with default settings |
| Database matching | ZINC15 | N/A | Used for novelty assessment |
Algorithms
- Transformer (encoder-decoder) via tensor2tensor library
- Beam search decoding (beam sizes 4 and 10)
- Needleman-Wunsch global alignment for protein sequence similarity (EMBOSS)
- SMINA for molecular docking
- RDKit for validity checking, property calculation, and canonicalization
Models
- 4 layers, 128 hidden size, 4 attention heads
- Character-level tokenization with 71-symbol vocabulary
- 5-fold Monte Carlo cross-validation with < 20% sequence similarity between train/test proteins
Evaluation
| Metric | Value | Notes |
|---|---|---|
| Valid SMILES | 90.2% (1-per-1), 82.6% (10-per-1) | Averaged across 5 splits |
| Unique SMILES | 92.3% (1-per-1), 81.7% (10-per-1) | Averaged across 5 splits |
| ZINC15 match | 30.6% (1-per-1), 17.1% (10-per-1) | Averaged across 5 splits |
| QED | 0.66 +/- 0.19 (1-per-1), 0.58 +/- 0.21 (10-per-1) | Drug-likeness score |
| SAS compliance | 99.9% (1-per-1), 100% (10-per-1) | SAS < 6 |
Hardware
- Google Colaboratory with one GPU
- Training for 600K epochs
Artifacts
| Artifact | Type | License | Notes |
|---|---|---|---|
| molecule_structure_generation | Code | Not specified | Jupyter Notebook implementation using tensor2tensor |
Paper Information
Citation: Grechishnikova, D. (2021). Transformer neural network for protein-specific de novo drug generation as a machine translation problem. Scientific Reports, 11, 321. https://doi.org/10.1038/s41598-020-79682-4
@article{grechishnikova2021transformer,
title={Transformer neural network for protein-specific de novo drug generation as a machine translation problem},
author={Grechishnikova, Daria},
journal={Scientific Reports},
volume={11},
number={1},
pages={321},
year={2021},
publisher={Nature Publishing Group},
doi={10.1038/s41598-020-79682-4}
}