A Language Model Approach to Structure-Based Drug Design
This is a Method paper that introduces Lingo3DMol, a pocket-based 3D molecule generation model combining transformer language models with geometric deep learning. The primary contribution is threefold: (1) a new molecular representation called FSMILES (fragment-based SMILES) that encodes both 2D topology and 3D spatial coordinates, (2) a dual-decoder architecture that jointly predicts molecular topology and atomic positions, and (3) an auxiliary non-covalent interaction (NCI) predictor that guides molecule generation toward favorable binding modes.
Limitations of Existing 3D Molecular Generative Models
Existing approaches to structure-based drug design fall into two categories, each with notable limitations. Graph-based autoregressive methods (e.g., Pocket2Mol) represent molecules as 3D graphs and use GNNs for generation, but frequently produce non-drug-like structures: large rings (seven or more atoms), honeycomb-like ring arrays, and molecules with either too many or too few rings. The autoregressive sampling process tends to get stuck in local optima early in generation and accumulates errors at each step. Diffusion-based methods (e.g., TargetDiff) avoid autoregressive generation but still produce a notable proportion of undesirable structures due to weak perception of molecular topology, since they do not directly encode or predict bonds. Both approaches struggle with metrics like QED (quantitative estimate of drug-likeness) and SAS (synthetic accessibility score), and neither reliably reproduces known active compounds when evaluated on protein pockets.
FSMILES: Fragment-Based SMILES with Dual Coordinate Systems
The core innovation of Lingo3DMol is a new molecular sequence representation called FSMILES that addresses the topology problem inherent in atom-by-atom generation. FSMILES reorganizes a molecule into fragments using a ring-first, depth-first traversal. Each fragment is represented using standard SMILES syntax, and the full molecule is assembled by combining fragments with a specific connection syntax. Ring size information is encoded directly in atom tokens (e.g., C_6 for a carbon in a six-membered ring), providing the autoregressive decoder with critical context about local topology before it needs to close the ring.
The model integrates two coordinate systems. Local spherical coordinates encode bond length ($r$), bond angle ($\theta$), and dihedral angle ($\phi$) relative to three reference atoms (root1, root2, root3). These are predicted using separate MLP heads:
$$r = \operatorname{argmax}\left(\operatorname{softmax}\left(\operatorname{MLP}_1\left(\left[E_{\text{type}}(\text{cur}), H_{\text{topo}}, h_{\text{root1}}\right]\right)\right)\right)$$
$$\theta = \operatorname{argmax}\left(\operatorname{softmax}\left(\operatorname{MLP}_2\left(\left[E_{\text{type}}(\text{cur}), H_{\text{topo}}, h_{\text{root1}}, h_{\text{root2}}\right]\right)\right)\right)$$
$$\phi = \operatorname{argmax}\left(\operatorname{softmax}\left(\operatorname{MLP}_3\left(\left[E_{\text{type}}(\text{cur}), H_{\text{topo}}, h_{\text{root1}}, h_{\text{root2}}, h_{\text{root3}}\right]\right)\right)\right)$$
Global Euclidean coordinates ($x, y, z$) are predicted by a separate 3D decoder ($D_{\text{3D}}$). During inference, the model defines a search space around the predicted local coordinates ($r \pm 0.1$ A, $\theta \pm 2°$, $\phi \pm 2°$) and selects the global position with the highest joint probability within that space. This fusion strategy exploits the rigidity of bond lengths and angles (which makes local prediction easier) while maintaining global spatial awareness.
NCI/Anchor Prediction Model
A separately trained NCI/anchor prediction model identifies potential non-covalent interaction sites and anchor points in the protein pocket. This model shares the transformer architecture of the generation model and is initialized from pretrained parameters. It predicts whether each pocket atom will form hydrogen bonds, halogen bonds, salt bridges, or pi-pi stacking interactions with the ligand, and whether it lies within 4 A of any ligand atom (anchor points). The predicted NCI sites serve two purposes: they are incorporated as input features to the encoder, and they provide starting positions for molecule generation (the first atom is placed within 4.5 A of a sampled NCI site).
Pretraining and Architecture
The model uses a denoising pretraining strategy inspired by BART. During pretraining on 12 million drug-like molecules, the model receives perturbed molecules (with 25% of atoms deleted, coordinates perturbed by $\pm 0.5$ A, and 25% of carbon element types corrupted) and learns to reconstruct the original structure. The architecture is transformer-based with graph structural information encoded through distance and edge vector bias terms in the attention mechanism:
$$A_{\text{biased}} = \operatorname{softmax}\left(\frac{QK^{\top}}{\sqrt{d_k}} + B_D + B_J\right)V$$
The overall loss combines FSMILES token prediction, absolute coordinate prediction, and local coordinate predictions ($r$, $\theta$, $\phi$) with their auxiliary counterparts:
$$L = L_{\text{FSMILES}} + L_{\text{abs-coord}} + L_r + L_\theta + L_\phi + L_{r,\text{aux}} + L_{\theta,\text{aux}} + L_{\phi,\text{aux}}$$
Fine-tuning is performed on 11,800 protein-ligand complex samples from PDBbind 2020, with the first three encoder layers frozen to prevent overfitting.
Evaluation on DUD-E with Drug-Likeness Filtering
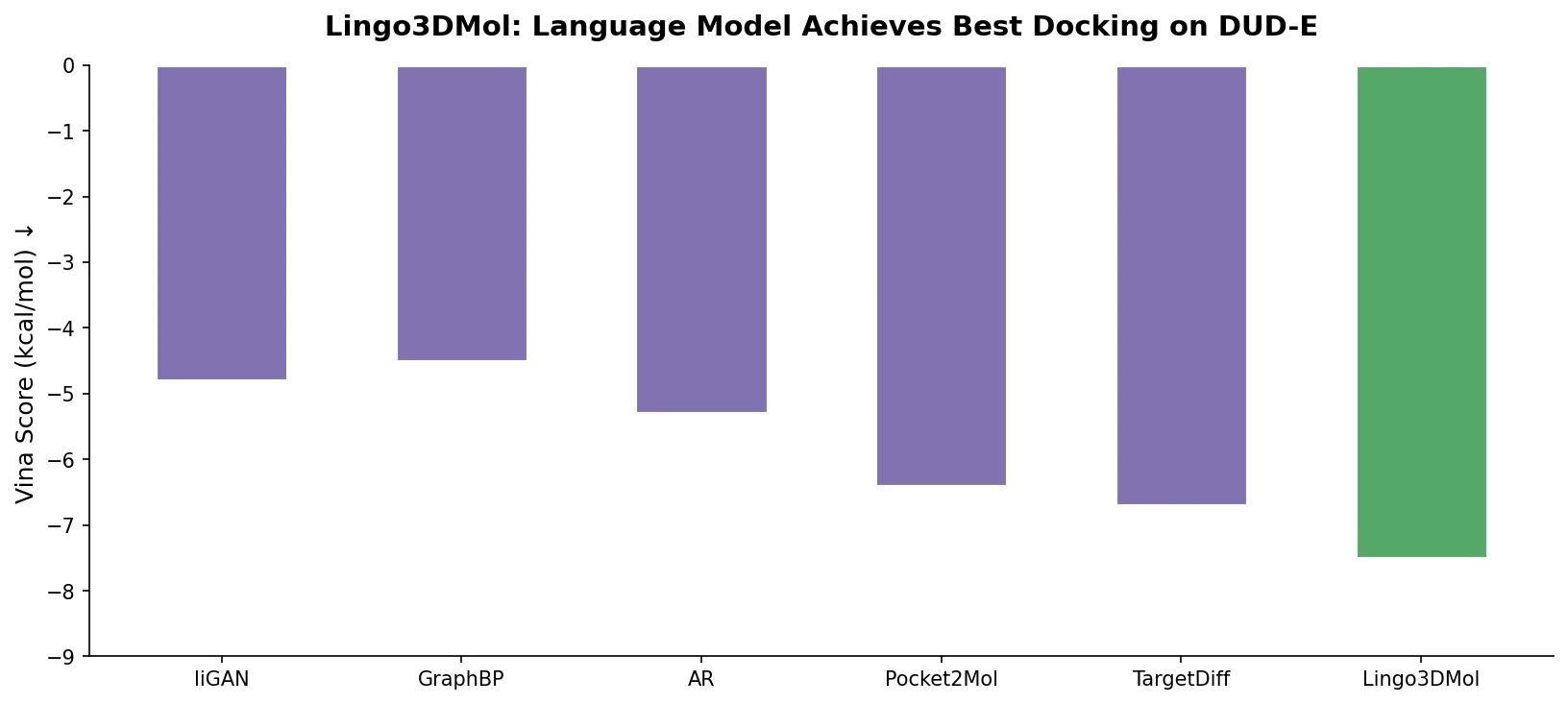
The evaluation uses the DUD-E dataset (101 targets, 20,000+ active compounds), comparing Lingo3DMol against Pocket2Mol and TargetDiff. A key methodological contribution is the emphasis on filtering generated molecules for drug-likeness (QED >= 0.3 and SAS <= 5) before evaluating binding metrics, as the authors demonstrate that molecules with good docking scores can still be poor drug candidates.
Molecular properties and binding mode (Table 1, drug-like molecules only):
| Metric | Pocket2Mol | TargetDiff | Lingo3DMol |
|---|---|---|---|
| Drug-like molecules (% of total) | 61% | 49% | 82% |
| Mean QED | 0.56 | 0.60 | 0.59 |
| Mean SAS | 3.5 | 4.0 | 3.1 |
| ECFP TS > 0.5 (% of targets) | 8% | 3% | 33% |
| Mean min-in-place GlideSP | -6.7 | -6.2 | -6.8 |
| Mean GlideSP redocking | -7.5 | -7.0 | -7.8 |
| Mean RMSD vs. low-energy conformer (A) | 1.1 | 1.1 | 0.9 |
| Diversity | 0.84 | 0.88 | 0.82 |
Lingo3DMol generates substantially more drug-like molecules (82% vs. 61% and 49%) and finds similar-to-active compounds for 33% of targets compared to 8% (Pocket2Mol) and 3% (TargetDiff). The model also achieves the best min-in-place GlideSP scores and lowest RMSD versus low-energy conformers, indicating higher quality binding poses and more realistic 3D geometries.
Molecular geometry: Lingo3DMol demonstrated the lowest Jensen-Shannon divergence for all atom-atom distance distributions and produced significantly fewer molecules with large rings (0.23% with 7-membered rings vs. 2.59% for Pocket2Mol and 11.70% for TargetDiff).
Information leakage analysis: The authors controlled for information leakage by excluding proteins with >30% sequence identity to DUD-E targets from training. When DUD-E targets were stratified by sequence identity to Pocket2Mol’s training set, Lingo3DMol’s advantage widened as leakage decreased, suggesting the performance gap is genuine rather than an artifact of training overlap.
Ablation studies (Table 2):
| Metric | Standard | Random NCI | No Pretraining |
|---|---|---|---|
| Drug-like (%) | 82% | 47% | 71% |
| ECFP TS > 0.5 | 33% | 6% | 3% |
| Mean min-in-place GlideSP | -6.8 | -5.8 | -4.9 |
| Dice score | 0.25 | 0.15 | 0.13 |
Both pretraining and the NCI predictor are essential. Removing pretraining reduces the number of valid molecules and binding quality. Replacing the trained NCI predictor with random NCI site selection severely degrades drug-likeness and the ability to generate active-like compounds.
Key Findings, Limitations, and Future Directions
Lingo3DMol demonstrates that combining language model sequence generation with geometric deep learning can produce drug-like 3D molecules that outperform graph-based and diffusion-based alternatives in binding mode quality, drug-likeness, and similarity to known actives. The FSMILES representation successfully constrains generated molecules to realistic topologies by encoding ring size information and using fragment-level generation.
Several limitations are acknowledged. Capturing all non-covalent interactions within a single molecule remains difficult with autoregressive generation. The model does not enforce equivariance (SE(3) invariance is approximated via rotation/translation augmentation and invariant features rather than built into the architecture). The pretraining dataset is partially proprietary (12M molecules from a commercial library, of which 1.4M from public sources are shared). Diversity of generated drug-like molecules is slightly lower than baselines, though the authors argue that baseline diversity explores chemical space away from known active regions. A comprehensive evaluation of drug-like properties beyond QED and SAS metrics is identified as an important next step.
Future directions include investigating electron density representations for molecular interactions, incorporating SE(3) equivariant architectures (e.g., GVP, Vector Neurons), and developing more systematic drug-likeness evaluation frameworks.
Reproducibility Details
Data
| Purpose | Dataset | Size | Notes |
|---|---|---|---|
| Pretraining | In-house commercial library | 12M molecules (1.4M public) | Filtered for drug-likeness; conformers via ConfGen |
| Fine-tuning | PDBbind 2020 (general set) | 11,800 samples (8,201 PDB IDs) | Filtered for <30% sequence identity to DUD-E targets |
| NCI labels | PDBbind 2020 | Same as fine-tuning | Labeled using ODDT for H-bonds, halogen bonds, salt bridges, pi-pi stacking |
| Evaluation | DUD-E | 101 targets, 20,000+ active compounds | Standard benchmark for structure-based drug design |
| Geometry evaluation | CrossDocked2020 | 100 targets | Used for bond length and atom distance distribution comparisons |
Algorithms
- Transformer-based encoder-decoder with graph structural bias terms (distance matrix $B_D$, edge vector matrix $B_J$)
- Denoising pretraining: 25% atom deletion, coordinate perturbation ($\pm 0.5$ A), 25% carbon element type corruption
- Depth-first search sampling with reward function combining model confidence and anchor fulfillment
- Fine-tuning: first three encoder layers frozen
- Local-global coordinate fusion during inference with search space: $r \pm 0.1$ A, $\theta \pm 2°$, $\phi \pm 2°$
Models
- Generation model: transformer encoder-decoder with dual decoders ($D_{\text{2D}}$ for topology, $D_{\text{3D}}$ for global coordinates)
- NCI/anchor prediction model: same architecture, initialized from pretrained parameters
- Pretrained, fine-tuned, and NCI model checkpoints available on GitHub and figshare
Evaluation
| Metric | Lingo3DMol | Best Baseline | Notes |
|---|---|---|---|
| Drug-like molecules (%) | 82% | 61% (P2M) | QED >= 0.3, SAS <= 5 |
| ECFP TS > 0.5 (% targets) | 33% | 8% (P2M) | Tanimoto similarity to known actives |
| Min-in-place GlideSP | -6.8 | -6.7 (P2M) | Lower is better |
| GlideSP redocking | -7.8 | -7.5 (P2M) | Lower is better |
| RMSD vs. low-energy conformer | 0.9 A | 1.1 A (both) | Lower is better |
| Generation speed (100 mol) | 874 +/- 401 s | 962 +/- 622 s (P2M) | NVIDIA Tesla V100 |
Hardware
- Inference benchmarked on NVIDIA Tesla V100 GPUs
- Generation of 100 valid molecules per target: 874 +/- 401 seconds
Artifacts
| Artifact | Type | License | Notes |
|---|---|---|---|
| Lingo3DMol | Code | GPL-3.0 | Inference code and model architecture |
| Model checkpoints | Model | GPL-3.0 | Pretraining, fine-tuning, and NCI checkpoints |
| Training data | Dataset | Not specified | Partial pretraining data (1.4M public molecules), fine-tuning complexes, evaluation molecules |
| Online service | Other | N/A | Web interface for molecule generation |
Paper Information
Citation: Feng, W., Wang, L., Lin, Z., Zhu, Y., Wang, H., Dong, J., Bai, R., Wang, H., Zhou, J., Peng, W., Huang, B., & Zhou, W. (2024). Generation of 3D molecules in pockets via a language model. Nature Machine Intelligence, 6(1), 62-73. https://doi.org/10.1038/s42256-023-00775-6
@article{feng2024generation,
title={Generation of 3D molecules in pockets via a language model},
author={Feng, Wei and Wang, Lvwei and Lin, Zaiyun and Zhu, Yanhao and Wang, Han and Dong, Jianqiang and Bai, Rong and Wang, Huting and Zhou, Jielong and Peng, Wei and Huang, Bo and Zhou, Wenbiao},
journal={Nature Machine Intelligence},
volume={6},
number={1},
pages={62--73},
year={2024},
publisher={Nature Publishing Group},
doi={10.1038/s42256-023-00775-6}
}