Target-Conditioned Molecular Generation via Transformer and MCTS
AlphaDrug is a Method paper that proposes a target-specific de novo molecular generation framework. The primary contribution is the combination of two components: (1) an Lmser Transformer (LT) that embeds protein-ligand context through hierarchical skip connections from encoder to decoder, and (2) a Monte Carlo tree search (MCTS) procedure guided by both the LT’s predicted probabilities and docking scores from the SMINA program. The method generates SMILES strings autoregressively, with each symbol selection informed by look-ahead search over potential binding affinities.
Bridging the Gap Between Molecular Generation and Protein Targeting
Most deep learning methods for de novo molecular generation optimize physicochemical properties (LogP, QED, SA) without conditioning on a specific protein target. Virtual screening approaches rely on existing compound databases and are computationally expensive. The few methods that do consider protein targets, such as LiGANN and the transformer-based approach of Grechishnikova (2021), show limited docking performance. The core challenge is twofold: the search space of drug-like molecules is estimated at $10^{60}$ compounds, and learning protein-ligand interaction patterns from sequence data is difficult because proteins and ligands have very different structures and sequence lengths.
AlphaDrug addresses these gaps by proposing a method that jointly learns protein-ligand representations and uses docking-guided search to navigate the vast chemical space.
Lmser Transformer and Docking-Guided MCTS
The key innovations are the Lmser Transformer architecture and the MCTS search strategy.
Lmser Transformer (LT)
The standard transformer for sequence-to-sequence tasks passes information from the encoder’s top layer to the decoder through cross-attention. AlphaDrug identifies an information transfer bottleneck: deep protein features from the encoder’s final layer must serve all decoder layers. Inspired by the Lmser (least mean squared error reconstruction) network, the authors add hierarchical skip connections from each encoder layer to the corresponding decoder layer.
Each decoder layer receives protein features at the matching level of abstraction through a cross-attention mechanism:
$$f_{ca}(Q_m, K_S, V_S) = \text{softmax}\left(\frac{Q_m K_S^T}{\sqrt{d_k}}\right) V_S$$
where $Q_m$ comes from the ligand molecule decoder and $(K_S, V_S)$ are passed through skip connections from the protein encoder. This allows different decoder layers to access different levels of protein features, rather than all layers sharing the same top-level encoding.
The multi-head attention follows the standard formulation:
$$\text{MultiHead}(Q, K, V) = \text{Concat}(H_1, \dots, H_h) W^O$$
$$H_i = f_{ca}(Q W_i^Q, K W_i^K, V W_i^V)$$
MCTS for Molecular Generation
The molecular generation process models SMILES construction as a sequential decision problem. At each step $\tau$, the context $C_\tau = {S, a_1 a_2 \cdots a_\tau}$ consists of the protein sequence $S$ and the intermediate SMILES string. MCTS runs a fixed number of simulations per step, each consisting of four phases:
Select: Starting from the current root node, child nodes are selected using a variant of the PUCT algorithm:
$$\tilde{a}_{\tau+t} = \underset{a \in A}{\arg\max}\left(Q(\tilde{C}_{\tau+t-1}, a) + U(\tilde{C}_{\tau+t-1}, a)\right)$$
where $Q(\tilde{C}, a) = W_a / N_a$ is the average reward and $U(\tilde{C}, a) = c_{puct} \cdot P(a | \tilde{C}) \cdot \sqrt{N_t} / (1 + N_t(a))$ is an exploration bonus based on the LT’s predicted probability.
The Q-values are normalized to $[0, 1]$ using the range of docking scores in the tree:
$$Q(\tilde{C}, a) \leftarrow \frac{Q(\tilde{C}, a) - \min_{m \in \mathcal{M}} f_d(S, m)}{\max_{m \in \mathcal{M}} f_d(S, m) - \min_{m \in \mathcal{M}} f_d(S, m)}$$
Expand: At a leaf node, the LT computes next-symbol probabilities and adds child nodes to the tree.
Rollout: A complete molecule is generated greedily using LT probabilities. Valid molecules are scored with SMINA docking; invalid molecules receive the minimum observed docking score.
Backup: Docking values propagate back up the tree, updating visit counts and cumulative rewards.
Training Objective
The LT is trained on known protein-ligand pairs using cross-entropy loss:
$$J(\Theta) = -\sum_{(S,m) \in \mathcal{D}} \sum_{\tau=1}^{L_m} \sum_{a \in \mathcal{A}} y_a \ln P(a \mid C_\tau(S, m))$$
MCTS is only activated during inference, not during training.
Experiments on Diverse Protein Targets
Dataset
The authors use BindingDB, filtered to 239,455 protein-ligand pairs across 981 unique proteins. Filtering criteria include: human proteins only, IC50 < 100 nM, molecular weight < 1000 Da, and single-chain targets. Proteins are clustered at 30% sequence identity using MMseqs2, with 25 clusters held out for testing (100 proteins), and the remainder split 90/10 for training (192,712 pairs) and validation (17,049 pairs).
Baselines
- T+BS10: Standard transformer with beam search (K=10) from Grechishnikova (2021)
- LT+BS10: The proposed Lmser Transformer with beam search
- LiGANN: 3D pocket-to-ligand shape generation via BicycleGAN
- SBMolGen: ChemTS-based method with docking constraints
- SBDD-3D: 3D autoregressive graph-based generation
- Decoys: Random compounds from ZINC database
- Known ligands: Original binding partners from the database
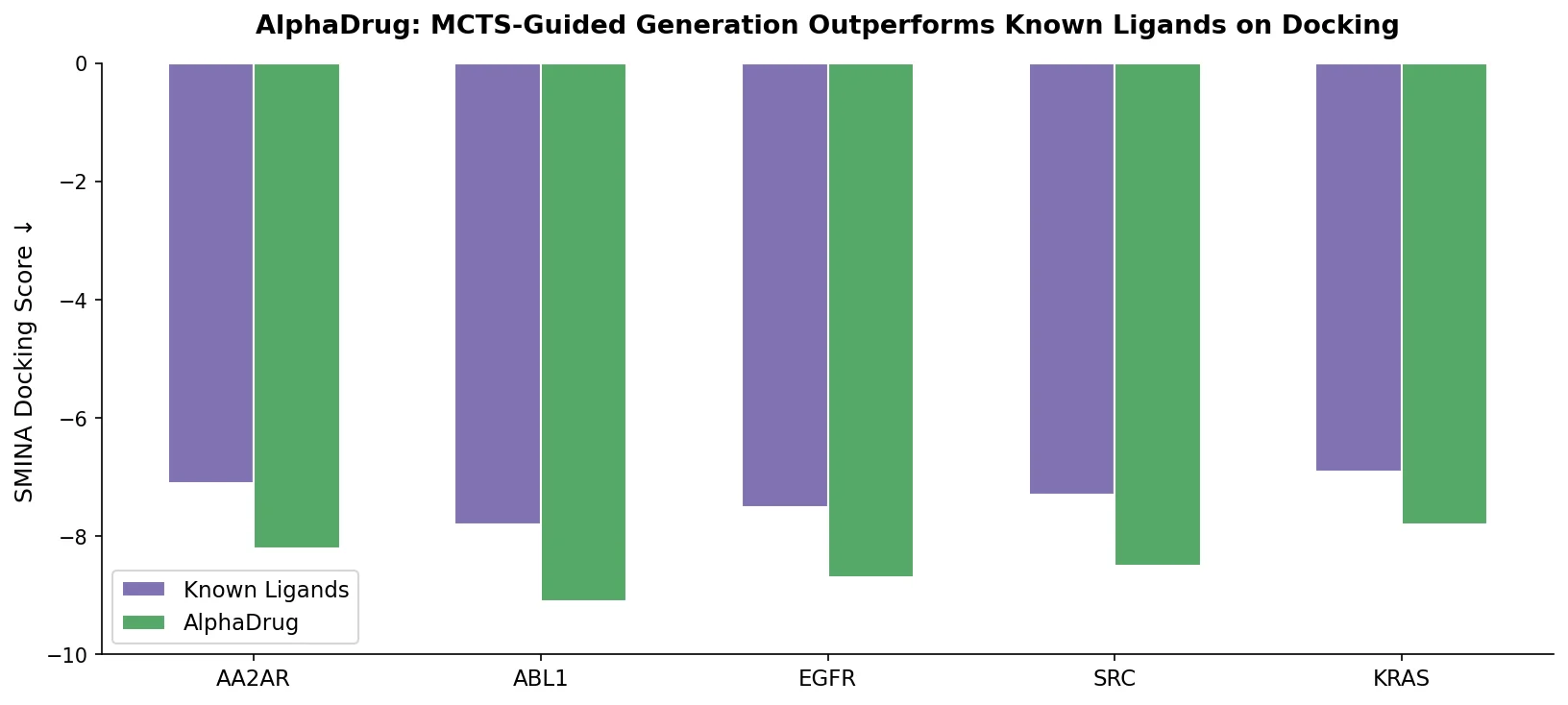
Main Results
| Method | Docking | Uniqueness | LogP | QED | SA | NP |
|---|---|---|---|---|---|---|
| Decoys | 7.3 | - | 2.4 | 0.8 | 2.4 | -1.2 |
| Known ligands | 9.8 | - | 2.2 | 0.5 | 3.3 | -1.0 |
| LiGANN | 6.7 | 94.7% | 2.9 | 0.6 | 3.0 | -1.1 |
| SBMolGen | 7.7 | 100% | 2.6 | 0.7 | 2.8 | -1.2 |
| SBDD-3D | 7.7 | 99.3% | 1.5 | 0.6 | 4.0 | 0.3 |
| T+BS10 | 8.5 | 90.6% | 3.8 | 0.5 | 2.8 | -0.8 |
| LT+BS10 | 8.5 | 98.1% | 4.0 | 0.5 | 2.7 | -1.0 |
| AlphaDrug (freq) | 10.8 | 99.5% | 4.9 | 0.4 | 2.9 | -1.0 |
| AlphaDrug (max) | 11.6 | 100% | 5.2 | 0.4 | 2.7 | -0.8 |
AlphaDrug (max) achieves the highest average docking score (11.6), surpassing known ligands (9.8). Statistical significance is confirmed with two-tailed t-test P-values below 0.01 for all comparisons.
MCTS vs. Beam Search Under Equal Compute
When constrained to the same number of docking evaluations, MCTS consistently outperforms beam search:
| Docking times (N) | BS | MCTS | P-value |
|---|---|---|---|
| N = 105 (S=10) | 8.4 (10.9) | 10.9 (11.5) | 1.8e-34 (4.5e-3) |
| N = 394 (S=50) | 8.3 (11.4) | 11.6 (12.2) | 1.4e-31 (1.8e-3) |
| N = 1345 (S=500) | 8.4 (11.9) | 12.4 (13.2) | 2.2e-39 (8.2e-6) |
Values in parentheses are average top-1 scores per protein.
Ablation: Effect of Protein Sequence Input
Replacing the full transformer (T) or LT with a transformer encoder only (TE, no protein input) demonstrates that protein conditioning improves both uniqueness and docking score per symbol (SpS):
| Method | Uniqueness | SpS | Molecular length |
|---|---|---|---|
| TE + MCTS (S=50) | 81.0% | 0.1926 | 62.93 |
| T + MCTS (S=50) | 98.0% | 0.2149 | 55.63 |
| LT + MCTS (S=50) | 100.0% | 0.2159 | 56.54 |
The SpS metric (docking score normalized by molecule length) isolates the quality improvement from the tendency of longer molecules to score higher.
Computational Efficiency
A docking lookup table caches previously computed protein-molecule docking scores, reducing actual docking calls by 81-86% compared to the theoretical maximum ($L \times S$ calls per molecule). With $S = 10$, AlphaDrug generates molecules in about 52 minutes per protein; with $S = 50$, about 197 minutes per protein.
Docking Gains with Acknowledged Limitations
Key Findings
- 86% of AlphaDrug-generated molecules have higher docking scores than known ligands for their respective targets.
- The LT architecture with hierarchical skip connections improves uniqueness (from 90.6% to 98.1% with beam search) and provides slight SpS gains over the vanilla transformer.
- MCTS is the dominant factor in performance improvement: even with only 10 simulations, it boosts docking scores by 31.3% over greedy LT decoding.
- Case studies on three proteins (3gcs, 3eig, 4o28) show that generated molecules share meaningful substructures with known ligands, suggesting chemical plausibility.
Limitations
The authors identify three areas for improvement:
- Sequence-only representation: AlphaDrug uses amino acid sequences rather than 3D protein structures. While it outperforms existing 3D methods (SBDD-3D), incorporating 3D pocket geometry could further improve performance.
- External docking as value function: SMINA docking calls are computationally expensive and become a bottleneck during MCTS. A learnable end-to-end value network would reduce this cost and allow joint policy-value training.
- Full rollout requirement: Every MCTS simulation requires generating a complete molecule for docking evaluation. Estimating binding affinity from partial molecules remains an open challenge.
The physicochemical properties (QED, SA) of AlphaDrug’s outputs are comparable to baselines but not explicitly optimized. LogP values trend toward the upper end of the Ghose filter range (4.9-5.2 vs. the 5.6 limit), which may indicate lipophilicity bias.
Reproducibility Details
Data
| Purpose | Dataset | Size | Notes |
|---|---|---|---|
| Training | BindingDB (filtered) | 192,712 protein-ligand pairs | Human proteins, IC50 < 100 nM, MW < 1000 Da |
| Validation | BindingDB (filtered) | 17,049 pairs | Same filtering criteria |
| Testing | BindingDB (filtered) | 100 proteins from 25 clusters | Clustered at 30% sequence identity via MMseqs2 |
Algorithms
- MCTS with PUCT selection criterion, $c_{puct} = 1.5$
- $S = 50$ simulations per step (default), $S = 10$ for fast variant
- Greedy rollout policy using LT probabilities
- Docking lookup table for efficiency (caches SMINA results)
- Two generation modes: max (deterministic, highest visit count) and freq (stochastic, proportional to visit counts)
Models
- Lmser Transformer with hierarchical encoder-to-decoder skip connections
- Sinusoidal positional encoding
- Multi-head cross-attention at each decoder layer
- Detailed hyperparameters (embedding dimensions, number of layers/heads) are in the supplementary material (Table S1)
Evaluation
| Metric | AlphaDrug (max) | Known ligands | Best baseline (T+BS10) |
|---|---|---|---|
| Docking score | 11.6 | 9.8 | 8.5 |
| Uniqueness | 100% | - | 90.6% |
| Validity | 100% | - | Not reported |
Hardware
Hardware specifications are not explicitly reported in the paper. Generation time is reported as approximately 52 minutes per protein ($S = 10$) and 197 minutes per protein ($S = 50$), with docking (via SMINA) being the dominant cost.
Artifacts
| Artifact | Type | License | Notes |
|---|---|---|---|
| CMACH508/AlphaDrug | Code | MIT | Official implementation, includes data processing and generation scripts |
Paper Information
Citation: Qian, H., Lin, C., Zhao, D., Tu, S., & Xu, L. (2022). AlphaDrug: protein target specific de novo molecular generation. PNAS Nexus, 1(4), pgac227. https://doi.org/10.1093/pnasnexus/pgac227
@article{qian2022alphadrug,
title={AlphaDrug: protein target specific de novo molecular generation},
author={Qian, Hao and Lin, Cheng and Zhao, Dengwei and Tu, Shikui and Xu, Lei},
journal={PNAS Nexus},
volume={1},
number={4},
pages={pgac227},
year={2022},
doi={10.1093/pnasnexus/pgac227}
}