Augmented Episodic Likelihood for Goal-Directed Generation
This is a Method paper that introduces REINVENT, a policy-based reinforcement learning framework for molecular de novo design. The primary contribution is a novel cost function, the augmented episodic likelihood, that fine-tunes a SMILES-based recurrent neural network (RNN) pre-trained on ChEMBL toward generating molecules satisfying user-defined property objectives. The method anchors the agent to the prior distribution of valid drug-like molecules, addressing failure modes of standard REINFORCE algorithms (reward exploitation and mode collapse to trivially simple structures).
De Novo Design Needs Flexible, Data-Driven Approaches
Traditional de novo design methods fall into three categories, each with limitations:
- Structure-based approaches grow ligands to fit binding pockets but often produce molecules with poor DMPK profiles and synthetic intractability.
- Ligand-based virtual library approaches generate large libraries and score them, but are constrained by pre-defined reaction rules or transformation rules that limit chemical diversity.
- Inverse QSAR methods attempt to map favorable activity regions back to molecular structures, but require descriptors suitable for both forward prediction and inverse mapping.
RNN-based generative models trained on SMILES offer a data-driven alternative that can learn the underlying distribution of drug-like chemical space without rigid rules. Segler et al. (2017) showed that fine-tuning a pre-trained RNN on focused actives yields high fractions of predicted actives. However, this maximum likelihood fine-tuning cannot use negative or continuous scores and risks catastrophic forgetting.
Prior RL approaches had significant issues. Jaques et al. (2016) used Deep Q-learning with prior likelihood regularization for sequence generation, but reported dependence on hand-written rules to penalize undesirable sequences and still observed reward exploitation producing unrealistically simple molecules. Standard REINFORCE algorithms tend to converge on trivial solutions (e.g., generating only “C” to satisfy a scoring function).
The Augmented Episodic Likelihood Framework
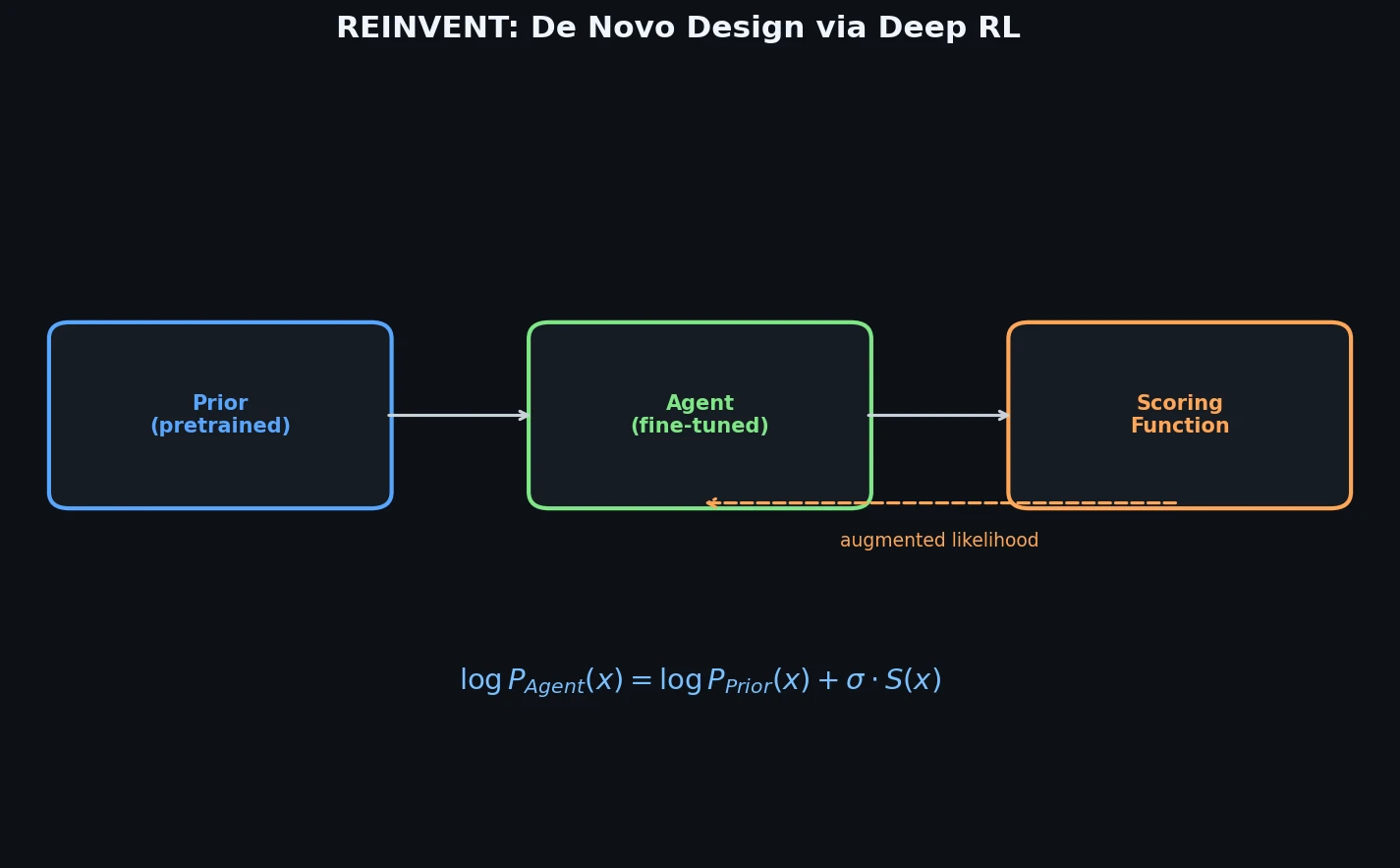
The core innovation is a formulation where the agent learns a policy that minimizes the squared difference between its own log-likelihood and an augmented target likelihood.
The RNN is first pre-trained on 1.5 million canonical SMILES from ChEMBL via maximum likelihood estimation:
$$ J(\Theta) = -\sum_{t=1}^{T} \log P(x^{t} \mid x^{t-1}, \dots, x^{1}) $$
The pre-trained model (the Prior) is then used as the starting point for the Agent. For a generated SMILES sequence $A = a_1, a_2, \dots, a_T$, the model likelihood is $P(A) = \prod_{t=1}^{T} \pi(a_t \mid s_t)$, and a scoring function $S(A) \in [-1, 1]$ rates desirability.
The augmented likelihood combines prior likelihood with the score:
$$ \log P(A)_{\mathbb{U}} = \log P(A)_{Prior} + \sigma S(A) $$
where $\sigma$ is a scalar coefficient controlling the trade-off between prior fidelity and score optimization.
The return is defined as the negative squared difference between the augmented likelihood and the agent’s likelihood:
$$ G(A) = -\left[\log P(A)_{\mathbb{U}} - \log P(A)_{\mathbb{A}}\right]^{2} $$
The agent minimizes $J(\Theta) = -G$, effectively learning a policy whose sequence likelihoods match the prior modulated by the scoring function. The authors show in supplementary material that this is equivalent to a REINFORCE algorithm with a specific final-step reward formulation.
This design has three key advantages over standard REINFORCE:
- The target policy is explicitly stochastic, preserving diversity in generated molecules
- The prior anchoring prevents catastrophic forgetting of SMILES syntax and chemical space coverage
- No hand-written rules are needed to penalize degenerate solutions
The Agent is trained on-policy with batches of 128 generated sequences, using SGD with learning rate 0.0005 and gradient clipping to $[-3, 3]$.
Three Experiments: Sulphur Avoidance, Celecoxib Analogues, and DRD2 Activity
Prior Network Architecture
The Prior is a 3-layer RNN with 1024 Gated Recurrent Units per layer, trained on RDKit canonical SMILES from ChEMBL (molecules with 10-50 heavy atoms, elements from ${H, B, C, N, O, F, Si, P, S, Cl, Br, I}$). Training used Adam ($\beta_1 = 0.9$, $\beta_2 = 0.999$, $\epsilon = 10^{-8}$) for 50,000 steps with batch size 128 and learning rate decay of 0.02 every 100 steps. The Prior generates 94% valid SMILES, of which 90% are novel.
Experiment 1: Learning to Avoid Sulphur
A proof-of-principle task where the scoring function assigns $S(A) = 1$ for valid sulphur-free molecules, $S(A) = 0$ for invalid SMILES, and $S(A) = -1$ for sulphur-containing molecules.
The Agent method was compared against three alternatives:
| Method | Fraction Valid | Fraction No S | Avg MW | Avg cLogP | Avg RotBonds | Avg AromRings |
|---|---|---|---|---|---|---|
| Prior | 0.94 | 0.66 | 371 | 3.36 | 5.39 | 2.26 |
| Agent | 0.95 | 0.98 | 367 | 3.37 | 5.41 | 2.26 |
| Action basis | 0.95 | 0.92 | 372 | 3.39 | 6.08 | 2.09 |
| REINFORCE | 0.98 | 0.98 | 585 | 11.3 | 30.0 | 0.57 |
| REINFORCE + Prior | 0.98 | 0.92 | 232 | 3.05 | 2.8 | 2.11 |
Standard REINFORCE exploited the reward by generating sequences of predominantly “C” (average MW 585, cLogP 11.3). REINFORCE + Prior avoided this but collapsed to small, simplistic structures (MW 232). The Agent achieved 98% sulphur-free structures while maintaining molecular properties nearly identical to the Prior, demonstrating that augmented episodic likelihood preserves the prior distribution.
Experiment 2: Similarity-Guided Generation (Celecoxib Analogues)
The scoring function uses Jaccard similarity on FCFP4 fingerprints:
$$ S(A) = -1 + 2 \times \frac{\min{J_{i,j}, k}}{k} $$
where $k$ caps the rewarded similarity. With $k = 1$ and $\sigma = 15$, the Agent recovers Celecoxib itself within 200 training steps. Even when all structures with $J > 0.5$ to Celecoxib (1,804 molecules) were removed from the Prior training set, the Agent still found Celecoxib after 400 steps, despite a 700-fold reduction in prior likelihood ($\log_e P$ from $-12.7$ to $-19.2$).
With moderate similarity targets ($k = 0.7$, $\sigma = 12$), the Agent generates diverse analogues including scaffold hops where functional groups are rearranged.
Experiment 3: Target Activity (DRD2)
The most drug-discovery-relevant task: generating molecules predicted active against the dopamine receptor type 2 (DRD2). An SVM classifier (Gaussian kernel, $C = 2^7$, $\gamma = 2^{-6}$) was trained on bioactivity data from ExCAPE-DB (7,218 actives with pIC50 > 5, 100,000 sampled inactives). The actives were split by Butina clustering (ECFP6, cutoff 0.4) to decrease nearest-neighbor similarity between train and test sets.
| Metric | Prior | Agent | Prior (reduced) | Agent (reduced) |
|---|---|---|---|---|
| Fraction valid SMILES | 0.94 | 0.99 | 0.94 | 0.99 |
| Fraction predicted actives | 0.03 | 0.97 | 0.02 | 0.96 |
| Fraction similar to train active | 0.02 | 0.79 | 0.02 | 0.75 |
| Fraction similar to test active | 0.01 | 0.46 | 0.01 | 0.38 |
| Test actives recovered (x10^-3) | 13.5 | 126 | 2.85 | 72.6 |
The Agent increased the fraction of predicted actives from 2-3% (Prior) to 96-97%, representing a 250-fold enrichment in the probability of generating a test set active. The Agent based on the reduced Prior (DRD2 actives removed from ChEMBL) still recovered 7% of test actives, meaning it generated experimentally confirmed actives that appeared in neither the generative model nor the activity prediction model training data.
Anchored Policy Learning Prevents Reward Exploitation
The key finding is that augmented episodic likelihood successfully balances score optimization with prior distribution preservation. The Agent achieves task objectives (sulphur avoidance, similarity targets, activity prediction) while maintaining the molecular property distributions learned from ChEMBL. This is a significant improvement over standard REINFORCE, which either exploits rewards trivially or collapses to simple structures.
Analysis of the conditional probability distributions between the Prior and Agent (for DRD2 active generation) shows that the policy changes are not drastic: most trends learned by the Prior carry over, with targeted modifications at specific steps that substantially alter sequence likelihoods and generated structure types.
Limitations acknowledged by the authors:
- All experiments use single-parameter scoring functions; multi-parametric optimization (activity + DMPK + synthetic accessibility) is left for future work
- The quality of generated structures depends heavily on the Prior’s coverage of chemical space
- The activity model (SVM) has limited domain of applicability, and structures outside this domain may be falsely scored
- No exhaustive study of how Prior training set size, model size, and regularization affect generation quality
Future directions include multi-parametric scoring functions, exploration of token embeddings, and adversarial training where the scoring function is replaced by a discriminator network (GAN-style training).
Reproducibility Details
Data
| Purpose | Dataset | Size | Notes |
|---|---|---|---|
| Prior training | ChEMBL | 1.5M structures | 10-50 heavy atoms, filtered elements |
| DRD2 activity model | ExCAPE-DB | 7,218 actives + 100K inactives | Butina clustering split (ECFP6, cutoff 0.4) |
| Similarity target | Celecoxib | 1 query structure | FCFP4 fingerprints for Jaccard similarity |
Algorithms
- Prior: 3-layer GRU RNN (1024 units/layer), Adam optimizer, 50K steps, batch size 128, LR 0.001 with 0.02 decay/100 steps
- Agent: Same architecture, SGD with LR 0.0005, gradient clipping [-3, 3], on-policy batches of 128
- DRD2 model: SVM with Gaussian kernel ($C = 2^7$, $\gamma = 2^{-6}$), grid search on validation set
Models
| Artifact | Type | License | Notes |
|---|---|---|---|
| REINVENT | Code | MIT | Original implementation in TensorFlow/Python 2.7 |
| Archived version | Code | MIT | Zenodo archive (DOI: 10.5281/zenodo.572576) |
Evaluation
- SMILES validity rate (RDKit parsing)
- Fraction of structures satisfying scoring function
- Molecular property distributions (MW, cLogP, rotatable bonds, aromatic rings)
- Jaccard similarity on ECFP6/FCFP4 fingerprints
- Recovery rate of known actives from test set
Hardware
Not specified in the paper. The implementation uses TensorFlow 1.0.1 with Python 2.7, RDKit, and Scikit-learn.
Paper Information
Citation: Olivecrona, M., Blaschke, T., Engkvist, O., & Chen, H. (2017). Molecular de-novo design through deep reinforcement learning. Journal of Cheminformatics, 9(1), 48.
@article{olivecrona2017molecular,
title={Molecular de-novo design through deep reinforcement learning},
author={Olivecrona, Marcus and Blaschke, Thomas and Engkvist, Ola and Chen, Hongming},
journal={Journal of Cheminformatics},
volume={9},
number={1},
pages={48},
year={2017},
publisher={Springer},
doi={10.1186/s13321-017-0235-x}
}