A Graph Recurrent Model for Molecular Generation with Property Optimization
This is a Method paper that introduces MolecularRNN, a graph-based recurrent generative model for molecular structures. The model extends GraphRNN to handle typed nodes (atom types) and typed edges (bond types), enabling direct generation of molecular graphs rather than working through string representations like SMILES. Three key contributions are combined: (1) the MolecularRNN architecture for autoregressive graph generation, (2) valency-based rejection sampling for guaranteed 100% validity at inference, and (3) policy gradient reinforcement learning for shifting molecular property distributions toward desired ranges.
Why Generate Molecules as Graphs Rather Than Strings
Computational de novo molecular design aims to create novel molecules with desired properties, a task central to drug discovery. At the time of this work, most deep generative models for molecules operated on SMILES strings, inheriting the complications of SMILES grammar and the problem that structurally similar molecules can have very different string representations. Graph-based representations are more natural for molecules, with atoms mapping to nodes and bonds to edges, and they allow direct enforcement of chemical constraints during generation.
Existing graph-based methods had their own limitations. Junction tree VAE (JT-VAE) generates molecules from structural fragments, which introduces ambiguity when converting junction trees back to molecules, particularly problematic during property optimization since molecules sharing a junction tree can have very different property values. The GCPN model uses graph convolutional networks with reinforcement learning but was evaluated only on top-3 generated molecules, making it difficult to assess overall distribution quality. Prior atom-level graph generation models like Li et al. (2018a) were restricted to molecules with at most 20 heavy atoms, limiting practical applicability.
Core Innovation: Extending GraphRNN with Chemical Constraints and RL
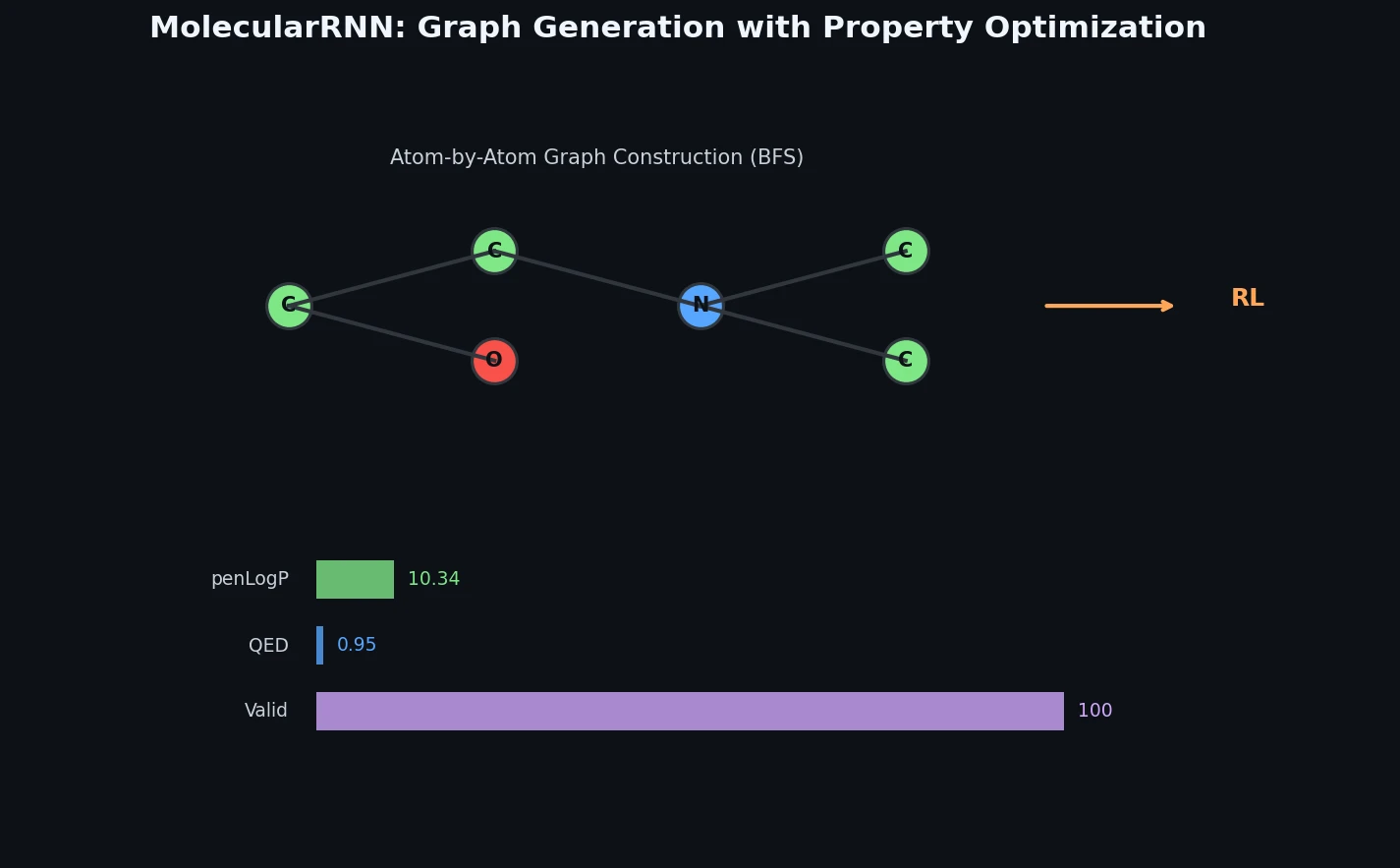
MolecularRNN builds on the GraphRNN architecture by introducing atom type predictions alongside edge type predictions. The model generates molecular graphs sequentially: at each step, a NodeRNN predicts the type of the next atom, then an EdgeRNN predicts bond types to all preceding atoms within a BFS-ordered window.
Autoregressive Graph Generation
The joint likelihood over atom types $C^{\pi}$ and adjacency vectors $S^{\pi}$ under BFS ordering $\pi$ is factorized as:
$$ p\left(S^{\pi}, C^{\pi}\right) = \prod_{i=1}^{n+1} p\left(C_{i}^{\pi} \mid S_{<i}^{\pi}, C_{<i}^{\pi}\right) p\left(S_{i}^{\pi} \mid C_{i}^{\pi}, S_{<i}^{\pi}, C_{<i}^{\pi}\right) $$
NodeRNN processes embeddings of previous atom types and adjacency vectors to produce a hidden state, from which a two-layer MLP with softmax predicts the next atom type $\psi_{i}$:
$$ h_{i}^{\text{node}} = \text{NodeRNN}\left(h_{i-1}^{\text{node}}, \left[\text{emb}(S_{i-1}^{\pi}), \text{emb}(C_{i-1}^{\pi})\right]\right) $$
$$ \psi_{i} = \text{NodeMLP}\left(h_{i}^{\text{node}}\right) $$
EdgeRNN then unrolls across preceding atoms to predict bond types $\phi_{i,j}$, initialized with the NodeRNN hidden state:
$$ h_{i,j}^{\text{edge}} = \text{EdgeRNN}\left(h_{i,j-1}^{\text{edge}}, \text{emb}(S_{i,j-1}^{\pi})\right), \quad h_{i,0}^{\text{edge}} = h_{i}^{\text{node}} $$
$$ \phi_{i,j} = \text{EdgeMLP}\left(h_{i,j}^{\text{edge}}\right) $$
Bond types are categorical over {no bond, single, double, triple}, and molecules are represented in kekulized form. BFS ordering limits the EdgeRNN window to $M = 12$ preceding atoms.
Valency-Based Rejection Sampling
During inference, each proposed bond of order $k$ between atoms $i$ and $j$ is accepted only if both atoms remain within their allowed valencies:
$$ \sum_{j} A_{i,j}^{\pi} + k \leq \text{valency}_{C_{i}^{\pi}} \quad \text{and} \quad \sum_{i} A_{i,j}^{\pi} + k \leq \text{valency}_{C_{j}^{\pi}} $$
Atoms that do not fill their valencies are complemented with hydrogens. This constraint can be enforced directly on graphs (unlike SMILES, where intermediate substrings are not chemically meaningful), yielding 100% valid molecules.
Property Optimization via Policy Gradient
For property optimization, MolecularRNN is formulated as a policy network in a Markov Decision Process. The loss function uses REINFORCE with a discounted final reward:
$$ L(\theta) = -\sum_{i=1}^{N} r(s_{N}) \cdot \gamma^{i} \cdot \log p(s_{i} \mid s_{i-1}; \theta) $$
where $r(s_{N})$ is the reward from a property critic and $\gamma$ is a discount factor. The authors also introduce a structural penalty during RL training that assigns a penalty of $-10$ to atoms violating valency constraints, providing a learning signal from invalid intermediate molecules.
Experimental Setup: Pretraining and Property Optimization
Pretraining
MolecularRNN is pretrained on three datasets: ChEMBL (~1.5M bioactive molecules), ZINC 250k (250K randomly selected commercially available compounds), and MOSES (~1.9M drug-like molecules from ZINC). The model considers 9 atom types (C, N, O, F, P, S, Cl, Br, I), 3 bond types (single, double, triple), and molecules with 10-50 heavy atoms. Architecture: NodeRNN with 4 GRU layers (hidden size 256), EdgeRNN with 4 GRU layers (hidden size 128), node embedding size 128, edge embedding size 16. Training uses Adam with learning rate 0.001 and multiplicative decay on 4 GPUs with batch size 512 per GPU for 250 epochs.
Generation Quality at Scale
The pretrained model generates 1 million molecules per dataset (far larger than prior work: JT-VAE used 5K samples, Li et al. used 100K). Results with valency-based rejection sampling:
| Training Set | Valid | Unique | Novel | IntDiv (p=1) | IntDiv (p=2) | SA Score | QED |
|---|---|---|---|---|---|---|---|
| ChEMBL | 100% | 99.2% | 99.3% | 0.895 | 0.890 | 3.67 +/- 1.20 | 0.56 +/- 0.20 |
| ZINC 250k | 100% | 99.8% | 100% | 0.892 | 0.887 | 3.60 +/- 1.01 | 0.68 +/- 0.16 |
| MOSES | 100% | 99.4% | 100% | 0.881 | 0.876 | 3.24 +/- 0.97 | 0.74 +/- 0.14 |
Comparison with baselines on ZINC 250k (30K samples):
| Method | Valid | Unique | Novel | SA Score | QED | IntDiv |
|---|---|---|---|---|---|---|
| JT-VAE | 99.8% | 100% | 100% | 3.37 | 0.76 | 0.85 |
| GCPN | 100% | 99.97% | 100% | 4.62 | 0.61 | 0.90 |
| MolecularRNN | 100% | 99.89% | 100% | 3.59 | 0.68 | 0.89 |
GCPN generates overly complex molecules (high SA score of 4.62), while MolecularRNN produces more realistic structures with higher internal diversity than JT-VAE.
Property Optimization Results
Policy gradient optimization is run for 300 iterations with batch size 512 and constant learning rate $10^{-5}$, discount factor $\gamma = 0.97$. Top-3 scores for penalized logP and QED:
| Method | logP 1st | logP 2nd | logP 3rd | QED 1st | QED 2nd | QED 3rd |
|---|---|---|---|---|---|---|
| ORGAN | 3.63 | 3.49 | 3.44 | 0.896 | 0.824 | 0.820 |
| JT-VAE | 5.30 | 4.93 | 4.49 | 0.925 | 0.911 | 0.910 |
| GCPN | 7.98 | 7.85 | 7.80 | 0.948 | 0.947 | 0.946 |
| MolecularRNN | 10.34 | 10.19 | 10.14 | 0.948 | 0.948 | 0.947 |
MolecularRNN achieves the highest penalized logP scores (10.34 vs. GCPN’s 7.98) while matching GCPN on QED. The authors also demonstrate melting temperature optimization using a GCN-based property predictor as the critic (RMSE 39.5 degrees C), showing that the RL framework generalizes to properties that cannot be computed directly from molecular graphs.
Distribution-Level Evaluation and Learned Chemical Patterns
The authors emphasize that reporting only top-3 scores is not informative, and they compare full property distributions. MolecularRNN shifts the QED distribution further toward maximum values compared to GCPN. They also note that during melting temperature optimization, the model rediscovered two chemical phenomena: fusing aromatic rings increases melting point, and the presence of polar groups (C=O, OH, NH2, heterocyclic nitrogens) enhances dipole-dipole interactions and raises melting temperature.
Without valency-based rejection sampling, the pretrained model achieves 65% validity. After structural penalty training (assigning -10 to valency-violating atoms and optimizing with policy gradient), validity increases to 90%. Enabling rejection sampling then achieves 100%.
Several limitations are worth noting. The BFS ordering introduces an arbitrary sequencing over equivalent graph traversals (the node order permutation problem is not addressed). The evaluation uses top-3 scores for property optimization, though the authors do advocate for distributional evaluation. The molecule size is capped at 50 heavy atoms. The paper does not report training time or wall-clock generation speed. Future directions mentioned include multi-objective property optimization and scaffold completion (graph completion from a given core structure).
Reproducibility Details
Data
| Purpose | Dataset | Size | Notes |
|---|---|---|---|
| Pretraining | ChEMBL | ~1.5M molecules | Bioactive molecules with experimental measurements |
| Pretraining | ZINC 250k | 250K molecules | Random subset of ZINC database |
| Pretraining | MOSES | ~1.9M molecules | Drug-like subset of ZINC |
| Melting point critic | Custom split | 37,940 train / 9,458 test | Melting temperatures from -196 to 517 degrees C |
Algorithms
- Pretraining: Maximum likelihood with Adam optimizer, learning rate 0.001 with multiplicative decay to $10^{-5}$, 250 epochs
- Structural penalty: Policy gradient with -10 penalty per valency-violating atom
- Property optimization: REINFORCE (policy gradient), 300 iterations, batch size 512, learning rate $10^{-5}$, discount factor $\gamma = 0.97$
- Melting point critic: GCN regression (4 layers, hidden size 128), Adam with learning rate 0.001, exponential decay $\gamma = 0.8$, 30 epochs, batch size 32
Models
- NodeRNN: 4 GRU layers, hidden size 256, node embedding 128
- EdgeRNN: 4 GRU layers, hidden size 128, edge embedding 16
- NodeMLP/EdgeMLP: 2-layer MLP with 128 hidden units, ReLU activation, softmax output
- BFS window: $M = 12$ preceding atoms
- Atom types: 9 (C, N, O, F, P, S, Cl, Br, I)
- Bond types: 3 (single, double, triple) + no bond
Evaluation
| Metric | Description |
|---|---|
| Validity | % chemically valid molecules (RDKit) |
| Uniqueness | % unique in generated pool (up to 1M) |
| Novelty | % not in training set |
| Internal Diversity | Average pairwise Tanimoto distance |
| SA Score | Synthetic accessibility (2-4 optimal range) |
| QED | Drug-likeness score (0-1) |
| Penalized logP | Lipophilicity with ring and SA penalties |
Hardware
- 4 GPUs (NVIDIA, specific model not stated)
- Per-GPU batch size of 512 for pretraining
- Training time not reported
Paper Information
Citation: Popova, M., Shvets, M., Oliva, J., & Isayev, O. (2019). MolecularRNN: Generating realistic molecular graphs with optimized properties. arXiv preprint arXiv:1905.13372.
@article{popova2019molecularrnn,
title={MolecularRNN: Generating realistic molecular graphs with optimized properties},
author={Popova, Mariya and Shvets, Mykhailo and Oliva, Junier and Isayev, Olexandr},
journal={arXiv preprint arXiv:1905.13372},
year={2019}
}