Multi-Objective De Novo Drug Design with Pareto Optimization
This is a Method paper that extends the DrugEx framework (v1) to handle multi-objective optimization in de novo drug design. The primary contribution is integrating Pareto-based ranking with evolutionary algorithm concepts (crossover and mutation) into an RNN-based reinforcement learning pipeline. The system generates SMILES-based molecules optimized simultaneously for activity toward multiple protein targets while avoiding off-targets, addressing polypharmacology scenarios where drugs must bind multiple specific receptors.
Polypharmacology and the Limits of Single-Objective Generation
Traditional drug discovery follows the “one drug, one target, one disease” paradigm, but drug molecules interact with an average of six protein targets. Off-target binding causes side effects that remain a leading cause of clinical failure and post-approval drug withdrawals (over 500 drugs withdrawn due to fatal toxicity). Complex diseases often require modulating multiple targets simultaneously, making polypharmacology an important design objective.
Prior deep learning approaches for de novo design, including DrugEx v1, focused on generating molecules active against a single target. Extending these methods to multiple objectives introduces fundamental challenges: objectives are often contradictory (high affinity for one target may correlate with high affinity for an undesired off-target), and naive weighted-sum approaches can collapse diversity by over-optimizing a single dominant objective. The authors specifically target the adenosine receptor system, where $A_1AR$ and $A_{2A}AR$ selectivity profiles matter for therapeutic efficacy, and hERG channel binding must be avoided to prevent cardiac toxicity.
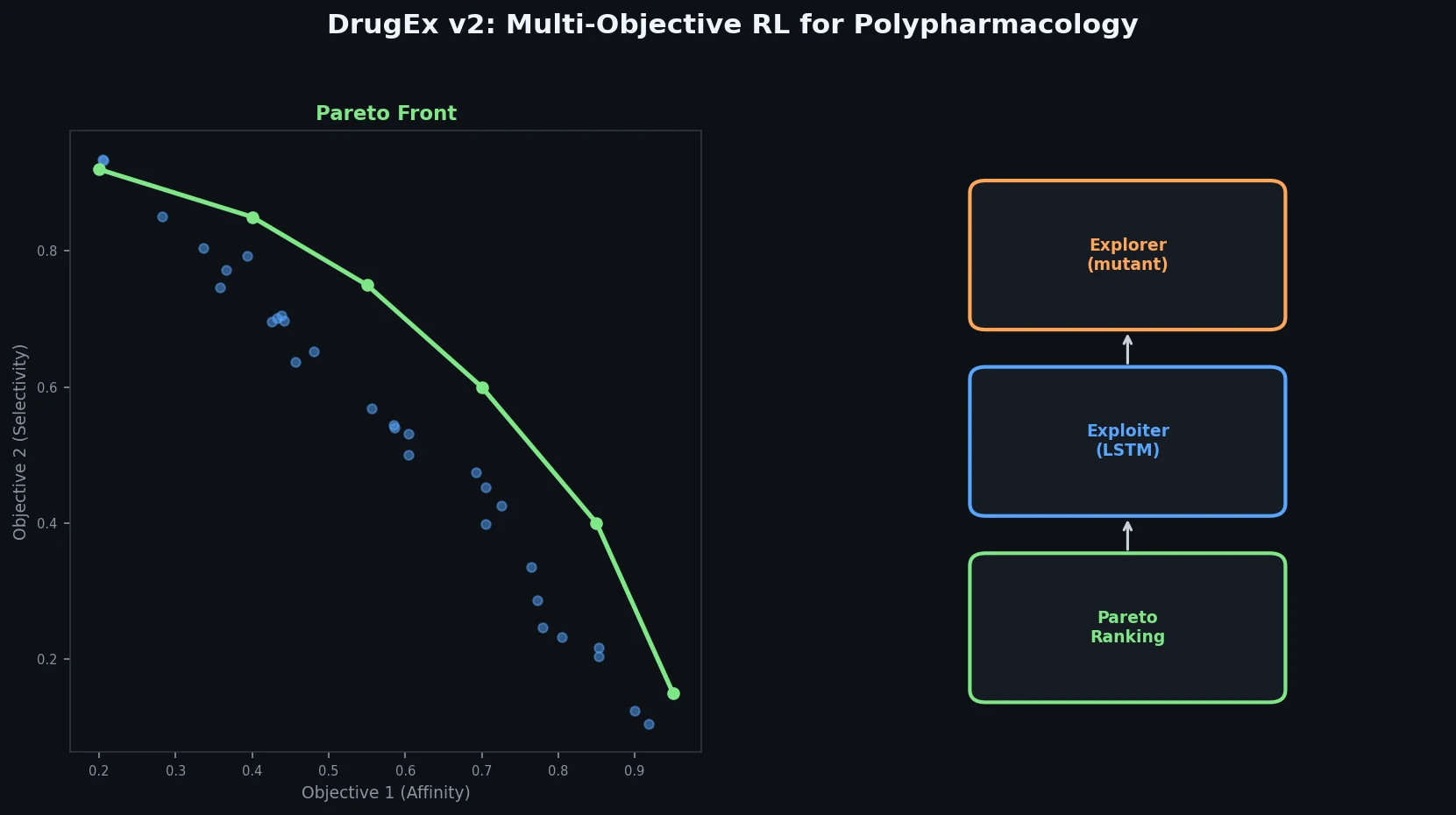
Evolutionary Exploration and Pareto Ranking in RL
The core innovation of DrugEx v2 has two components: an evolutionary exploration strategy and Pareto-based reward assignment.
Evolutionary Exploration Strategy
The generation process uses three RNN networks with identical LSTM architectures:
- Agent net ($G_A$): the primary generator, updated at each training epoch via policy gradient
- Crossover net ($G_C$): initialized from the fine-tuned model, updated iteratively from $G_A$ after each convergence period
- Mutation net ($G_M$): initialized from the pre-trained model, parameters fixed throughout training
At each token-generation step, a random number determines whether the token probability comes from the combination of $G_A$ and $G_C$ (with probability $1 - \varepsilon$) or from $G_M$ (with probability $\varepsilon$). This mirrors crossover and mutation operations from evolutionary algorithms, maintaining diversity while steering toward desired properties.
Pareto Front Reward Scheme
For $n$ objectives (three in this study: $A_1AR$, $A_{2A}AR$, hERG), each molecule receives a score $R_i$ based on its predicted bioactivity:
$$ R_{i} = \begin{cases} \text{minmax}(pX_{i}), & \text{if high affinity required} \\ 1 - \text{minmax}(pX_{i}), & \text{if low affinity required} \\ 0, & \text{if SMILES invalid} \end{cases} $$
where $pX_i$ is the predicted bioactivity (range 3.0 to 10.0), normalized to [0, 1].
For the multi-target case, high affinity is required for both $A_1AR$ and $A_{2A}AR$ while low affinity is required for hERG. For the target-specific case, high affinity is required only for $A_{2A}AR$ while low affinity is required for both $A_1AR$ and hERG.
Molecules are ranked using a non-dominated sorting algorithm to construct Pareto fronts. Within each front, molecules are ranked by average Tanimoto distance (using ECFP6 fingerprints) rather than crowding distance, favoring chemically diverse solutions. The final reward is:
$$ R_i^{*} = \begin{cases} 0.5 + \frac{k - N_{undesired}}{2N_{desired}}, & \text{if desired} \\ \frac{k}{2N_{undesired}}, & \text{if undesired} \end{cases} $$
where $k$ is the molecule’s index in the Pareto rank. Rewards for undesired and desired solutions are distributed in $(0, 0.5]$ and $(0.5, 1.0]$, respectively.
The agent is trained via policy gradient:
$$ J(\theta) = \mathbb{E}\left[R^{*}(y_{1:T}) \middle|\theta\right] = \sum_{t=1}^{T} \log G(y_t | y_{1:t-1}) \cdot R^{*}(y_{1:T}) $$
Weighted Sum Alternative
The authors also implement a weighted sum (WS) scheme with dynamic weights proportional to the ratio of undesired to desired molecules per objective:
$$ w_i = \frac{r_i}{\sum_{k=1}^{M} r_k}, \quad R^{*} = \sum_{i=1}^{n} w_i R_i $$
This auto-adjusts importance toward under-performing objectives during training.
Molecular Diversity Metric
Diversity is measured using the Solow-Polasky metric adapted from ecological biodiversity:
$$ I(A) = \frac{1}{|A|} \mathbf{e}^{\top} F(\mathbf{s})^{-1} \mathbf{e} $$
where $F(\mathbf{s})$ is a distance matrix with entries $f(d_{ij}) = e^{-\theta d_{ij}}$ and $d_{ij}$ is the Tanimoto distance between ECFP6 fingerprints of molecules $s_i$ and $s_j$.
Multi-Target and Target-Specific Experiments
QSAR Environment
Four ML algorithms were benchmarked for the bioactivity prediction environment: Random Forest (RF), SVM, PLS, and Multi-task DNN (MT-DNN). Input features combined 2048-bit ECFP6 fingerprints with 19 physicochemical descriptors (2067D total). The training data came from ChEMBL v26: 25,731 ligands with bioactivity measurements toward $A_1AR$, $A_{2A}AR$, and hERG. RF was selected as the final predictor based on superior performance in temporal-split independent testing ($R^2$ and RMSE), prioritizing robustness over cross-validation metrics.
Generative Model Architecture
The RNN generator uses six layers: input, embedding (128D), three LSTM recurrent layers (512 hidden units), and output. LSTM was chosen over GRU based on higher valid SMILES rates (97.5% vs. 93.1% for pre-trained, 97.9% vs. 95.7% for fine-tuned). Pre-training used 1.7M molecules from ChEMBL; fine-tuning used the 25,731 LIGAND set molecules.
Baselines
DrugEx v2 was compared against DrugEx v1, REINVENT, and ORGANIC, all using the same RNN architecture and pre-trained/fine-tuned models, with only the RL framework differing. Both Pareto front (PF) and weighted sum (WS) reward schemes were tested.
Multi-Target Results
In the multi-target case (high affinity for $A_1AR$ and $A_{2A}AR$, low affinity for hERG):
| Method | Scheme | Validity | Desirability | Uniqueness | Diversity |
|---|---|---|---|---|---|
| DrugEx v2 | PF | 99.57% | 80.81% | 87.29% | 0.70 |
| DrugEx v2 | WS | 99.80% | 97.45% | 89.08% | 0.49 |
| REINVENT | PF | 99.54% | 57.43% | 98.84% | 0.77 |
| ORGANIC | PF | 98.84% | 66.01% | 82.67% | 0.65 |
| DrugEx v1 | PF | 98.28% | 43.27% | 88.96% | 0.71 |
DrugEx v2 achieved the highest desirability under both schemes. The WS scheme maximized desirability (97.45%) but at the cost of diversity (0.49). The PF scheme maintained higher diversity (0.70) with still-strong desirability (80.81%).
Target-Specific Results
In the target-specific case (high $A_{2A}AR$, low $A_1AR$ and hERG):
| Method | Scheme | Validity | Desirability | Uniqueness | Diversity |
|---|---|---|---|---|---|
| DrugEx v2 | PF | 99.53% | 89.49% | 90.55% | 0.73 |
| DrugEx v2 | WS | 99.62% | 97.86% | 90.54% | 0.31 |
| REINVENT | WS | 99.55% | 81.27% | 98.87% | 0.34 |
| ORGANIC | PF | 98.29% | 86.98% | 80.30% | 0.64 |
DrugEx v2 with PF achieved high desirability (89.49%) while maintaining diversity (0.73), outperforming both the WS scheme’s diversity collapse (0.31) and competing methods.
Chemical Space Coverage
t-SNE visualization with ECFP6 descriptors showed that the PF scheme guided generators to cover chemical space more broadly than the WS scheme. DrugEx v1 and v2 covered nearly all of the chemical space occupied by known active ligands, while REINVENT and ORGANIC covered only partial regions in the target-specific case.
Substructure Distribution
Generated molecules were evaluated for purine ring, furan ring, and benzene ring frequencies. DrugEx v2 with PF produced substructure distributions closest to the LIGAND set, suggesting it better preserves the chemical characteristics of known active molecules compared to REINVENT (which over-represented benzene rings) and ORGANIC.
GuacaMol Benchmark
DrugEx v2 was tested on 20 goal-directed tasks from the GuacaMol benchmark, achieving the best score in 12 of 20 tasks and an overall second place. The method struggled with tasks requiring contradictory objectives in narrow chemical spaces (e.g., the Sitagliptin MPO task), reflecting its emphasis on diverse feasible molecules rather than optimal individual solutions.
Diversity-Desirability Trade-off and Limitations
The key finding is that the Pareto front scheme and weighted sum scheme offer complementary strengths: PF produces molecules with higher diversity and more realistic substructure distributions, while WS achieves higher raw desirability scores. The Pareto front scheme is preferred for polypharmacology applications where chemical diversity matters for lead optimization.
The mutation rate $\varepsilon$ controls the diversity-desirability trade-off. Higher $\varepsilon$ increases diversity at the cost of desirability. The authors tested $\varepsilon \in {10^{-2}, 10^{-3}, 10^{-4}, 0}$ and found that appropriate tuning is important.
Limitations acknowledged by the authors include:
- The method is less effective for tasks with contradictory objectives in narrow chemical spaces
- Emphasis is on generating diverse feasible molecules rather than individual optimal solutions
- REINVENT 2.0 did not converge with the PF scheme, suggesting the Pareto approach may not be universally compatible with all RL frameworks
- Bioactivity predictions rely on QSAR models (RF), which may not generalize perfectly to novel chemical scaffolds
Future directions mentioned include adopting newer architectures (BERT, Transformer, GPT-2), handling graph and fragment representations, and integrating additional objectives like stability and synthesizability.
Reproducibility Details
Data
| Purpose | Dataset | Size | Notes |
|---|---|---|---|
| Pre-training | ChEMBL v26 (ChEMBL set) | 1.7M molecules | SMILES syntax learning, drug-like molecules |
| Fine-tuning / Environment | LIGAND set | 25,731 ligands | Bioactivities for $A_1AR$, $A_{2A}AR$, hERG from ChEMBL |
| Benchmark | GuacaMol | 20 tasks | Goal-directed generation tasks |
Active/inactive thresholds: $pX \geq 6.5$ (active), $pX < 6.5$ (inactive). Low-quality data without exact pX assigned $pX = 3.99$ with sample weight 0.1.
Algorithms
- QSAR predictor: Random Forest, 1000 trees, Gini criterion. Input: 2048-bit ECFP6 + 19 physicochemical properties (2067D). MinMax normalization.
- Generator: 6-layer RNN with LSTM cells (512 hidden units), embedding dim 128, vocabulary 84 tokens. Adam optimizer, lr $10^{-3}$, batch size 512, 1000 epochs.
- RL training: Policy gradient with Pareto-based or weighted-sum reward. Mutation rates tested: $\varepsilon \in {10^{-2}, 10^{-3}, 10^{-4}, 0}$.
- Pareto ranking: GPU-accelerated non-dominated sorting via PyTorch. Tanimoto-based crowding distance with ECFP6 fingerprints.
Models
| Component | Architecture | Parameters |
|---|---|---|
| Generator | LSTM (3 layers, 512 hidden) | Embedding 128D, vocab 84 |
| Predictor | Random Forest | 1000 trees, 2067D input |
| MT-DNN (alternative) | 3 hidden layers (4000, 2000, 1000) | ReLU, 20% dropout |
Evaluation
| Metric | Description |
|---|---|
| Validity | Fraction of generated SMILES that parse to valid molecules |
| Desirability | Fraction of molecules meeting all activity thresholds ($pX \geq 6.5$ on-targets, $pX < 6.5$ off-targets) |
| Uniqueness | Fraction of non-duplicate molecules |
| Diversity | Solow-Polasky metric on ECFP6 Tanimoto distances |
| SA score | Synthetic accessibility (1-10, lower is easier) |
| QED | Quantitative estimate of drug-likeness (0-1, higher is better) |
Hardware
GPU acceleration was used for Pareto optimization via PyTorch. Specific hardware details (GPU model, training time) are not reported in the paper.
Artifacts
| Artifact | Type | License | Notes |
|---|---|---|---|
| DrugEx GitHub | Code | MIT | Official implementation (Python, PyTorch) |
| ChEMBL v26 | Dataset | CC BY-SA 3.0 | Source of training molecules and bioactivity data |
Paper Information
Citation: Liu, X., Ye, K., van Vlijmen, H. W. T., Emmerich, M. T. M., IJzerman, A. P., & van Westen, G. J. P. (2021). DrugEx v2: de novo design of drug molecules by Pareto-based multi-objective reinforcement learning in polypharmacology. Journal of Cheminformatics, 13(1), 85. https://doi.org/10.1186/s13321-021-00561-9
@article{liu2021drugex,
title={DrugEx v2: de novo design of drug molecules by Pareto-based multi-objective reinforcement learning in polypharmacology},
author={Liu, Xuhan and Ye, Kai and van Vlijmen, Herman W. T. and Emmerich, Michael T. M. and IJzerman, Adriaan P. and van Westen, Gerard J. P.},
journal={Journal of Cheminformatics},
volume={13},
number={1},
pages={85},
year={2021},
doi={10.1186/s13321-021-00561-9}
}