Curriculum Learning as a Method for Molecular Generation
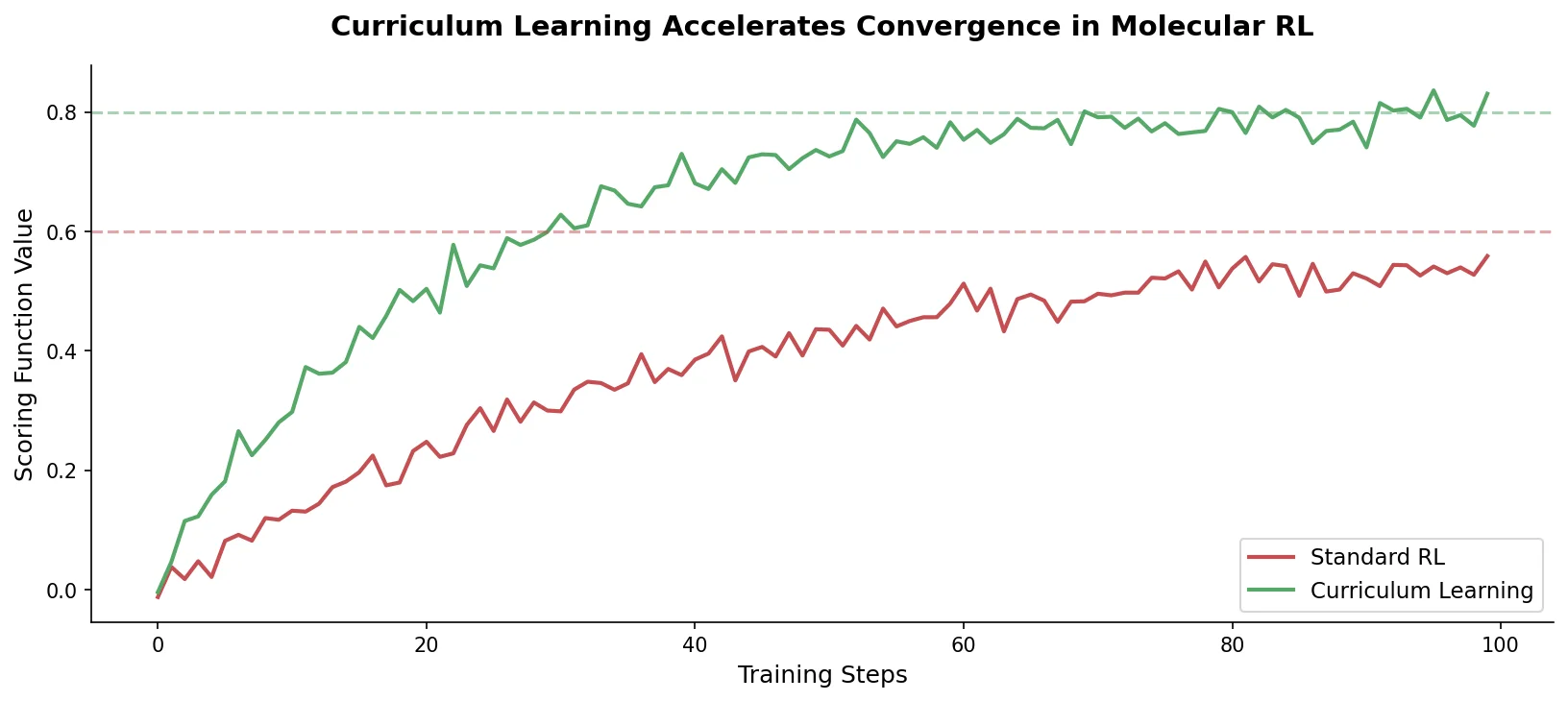
This is a Method paper that introduces curriculum learning (CL) into the REINVENT de novo molecular design platform. The primary contribution is a training strategy that decomposes complex multi-parameter optimization (MPO) objectives into sequences of simpler tasks with increasing complexity. The agent learns each simpler task before progressing to the full production objective, accelerating convergence and improving the quality and diversity of generated molecules compared to standard policy-based reinforcement learning (RL).
The Computational Cost of Complex Reward Functions
Policy-based RL for molecular design works by training a generative model (the agent) to produce molecules that maximize a reward function. In practice, drug design reward functions often include computationally expensive components such as molecular docking. When the reward landscape is complex and minima are difficult to find, the agent may spend many epochs sampling molecules far from the desired objective. The resulting small gradients cause minimal policy updates, leading to long periods of non-productivity. This is particularly wasteful when each reward evaluation involves expensive physics-based computations.
The core problem is that standard RL treats the full MPO objective as a monolithic task. If the agent cannot find any rewarding molecules early in training, it receives near-zero gradients and makes negligible progress. This creates a bootstrapping problem: the agent needs to already be sampling from favorable regions of chemical space to receive useful learning signals, but it has no guidance on how to get there.
Curriculum learning, originally proposed by Bengio et al. (2009), addresses this by arranging training tasks in order of increasing difficulty. When constituent tasks are correlated with the final objective, the gradients from simpler tasks provide more effective traversal of the optimization landscape.
Formalized Curriculum Strategy for REINVENT
The key innovation is a two-phase training protocol with formal definitions for curriculum progression.
A scoring function maps SMILES strings to desirability scores in $[0, 1]$ using a weighted geometric mean:
$$S(x) = \left(\prod_{i=1}^{n} c_{i}(x)^{w_{i}}\right)^{1 / \sum_{i=1}^{n} w_{i}}$$
where $x$ is a sampled compound in SMILES format, $c_{i}$ is the $i$-th scoring component, and $w_{i}$ is its weight.
A Curriculum $C$ consists of a sequence of Objectives $O = {O_{C_1}, \ldots, O_{C_n}, O_{P}}$, where subscripts $C$ and $P$ denote Curriculum and Production Objectives respectively. Each Objective has a corresponding scoring function. Progression is controlled by Curriculum Progression Criteria $P = {P_{1}, \ldots, P_{n}}$, where each $P_{i}$ defines a score threshold the agent must achieve before advancing to the next objective.
Curriculum Phase: The agent trains on sequential Curriculum Objectives with increasing complexity. A diversity filter is not applied during this phase, as it could be counterproductive to guiding the agent toward favorable chemical space. No computationally expensive components (e.g., docking) are used in Curriculum Objectives.
Production Phase: Activated only when the final Curriculum Progression Criterion $P_{n}$ is satisfied. The agent now optimizes the full Production Objective, which may include expensive components like molecular docking. A new inception memory is initialized (clearing Curriculum Phase compounds), and a Bemis-Murcko scaffold diversity filter is applied to encourage exploration across multiple local minima.
The implementation builds on REINVENT’s RNN architecture: three hidden layers of 512 LSTM cells with an embedding size of 256 and a linear layer with softmax activation, pretrained on ChEMBL to learn SMILES syntax.
Three Experiments on PDK1 Inhibitor Design
The authors evaluate CL on three molecular design tasks of increasing complexity, all centered on designing 3-phosphoinositide-dependent protein kinase-1 (PDK1) inhibitors.
Experiment 1: Target Scaffold Construction
The goal is to generate compounds possessing a dihydro-pyrazoloquinazoline scaffold with a phenyl substituent, a scaffold not present in the prior’s training set. Standard RL fails entirely over 2000 epochs because the probability of randomly sampling a compound with this scaffold is negligibly small, producing binary rewards (1.0 if scaffold present, 0.5 otherwise) that never rise above 0.5.
CL decomposes the target scaffold into 5 progressively complex substructures. Each Curriculum Progression Criterion threshold is set to 0.8. The agent learns to generate compounds with each substructure before advancing. CL finds the target scaffold within 1750 epochs, while baseline RL cannot find it in the same timeframe.
Experiments 2 and 3: Molecular Docking Constraints
These experiments use a Production Objective combining a molecular docking constraint (retaining two hydrogen-bonding interactions with Ala 162 of PDK1, PDB ID: 2XCH) and QED (Quantitative Estimate of Druglikeness). Both experiments limit computational cost by capping production epochs at 300.
Experiment 2 uses Tanimoto (2D) similarity to a reference ligand as the Curriculum Objective. Two scenarios are tested: “Low” (threshold 0.5) and “High” (threshold 0.8).
Experiment 3 uses ROCS (3D) shape-based similarity to the reference ligand as the Curriculum Objective, with “Low” (0.5) and “High” (0.75) thresholds.
All experiments are run in triplicate. The baseline includes both standard RL and RL with Tanimoto/ROCS components added directly to the scoring function (not sequentially), to control for the presence of these components.
Across all CL experiments, CL generates between 2,941 and 9,068 more compounds with docking scores better than the reference ligand (-10.907 kcal/mol) compared to baseline RL, corresponding to 12.42-23.79% improvement in the fraction of compounds exceeding the reference. Between the Curriculum Objectives, the “High” threshold scenario outperforms the “Low” scenario by 316-3,415 additional compounds (with percentage improvements ranging from -0.4% to 10.57%).
Baseline RL produces essentially no compounds satisfying the docking constraint for the first 100 epochs. CL agents achieve immediate productivity: in the “High” Tanimoto scenario, the initial docking score already exceeds the maximum score achieved by baseline RL over 300 epochs.
Scaffold Diversity Analysis
CL generates more unique Bemis-Murcko scaffolds than baseline RL in all experiments. The “High” scenarios produce more unique scaffolds than the “Low” scenarios. CL also produces a higher fraction of “favorable” scaffolds (those with better docking scores than the reference ligand).
Accelerated Convergence with a Diversity Trade-off
The results demonstrate three consistent findings across all experiments:
Accelerated productivity: CL agents reach productive sampling of favorable compounds substantially faster than baseline RL. Even a single Curriculum Objective with a computationally inexpensive metric can guide the agent to regions of chemical space where expensive Production Objectives are readily satisfied.
Improved output quality: CL generates more compounds with favorable docking scores, more unique scaffolds, and a higher fraction of scaffolds that outperform the reference ligand.
Controllable trade-off: The Curriculum Progression Criterion threshold provides direct control over agent policy. Higher thresholds produce better Production Objective optimization but reduce intra-set diversity (higher cross-Tanimoto similarities among generated compounds). UMAP visualizations confirm that “Low” and “High” scenarios sample from nearby but distinct regions of chemical space.
The authors note that even moderate optimization of similarity-based Curriculum Objectives (the “Low” scenarios) already substantially narrows the agent’s perceived solution space. This suggests that CL inherently regularizes the agent policy, trading some diversity for convergence speed.
Limitations: The authors acknowledge that data supporting the findings are available only upon request rather than as public deposits. The approach is demonstrated on a single target (PDK1), and the curriculum design requires domain expertise to decompose objectives appropriately. The inverse relationship between Curriculum Objective optimization and solution diversity means practitioners must carefully tune thresholds for their specific applications.
Reproducibility Details
Data
| Purpose | Dataset | Size | Notes |
|---|---|---|---|
| Prior training | ChEMBL | Not specified | Used to pretrain the RNN on SMILES syntax |
| Docking target | PDB 2XCH | 1 structure | PDK1 receptor crystal structure |
Raw data supporting the findings are available from the corresponding author upon request.
Algorithms
- REINVENT platform with LSTM-based RNN (3 hidden layers, 512 cells, embedding size 256)
- Scoring function: weighted geometric mean of components
- Curriculum Progression Criteria: score thresholds (0.5 or 0.75-0.8 depending on scenario)
- Diversity filter: Identical Murcko Scaffold with bucket size 25 (Production Phase only)
- Inception (experience replay) for both phases, reset at phase transition
- Batch size: 128, learning rate: 0.0001, sigma: 128, Adam optimizer
Models
- Prior: RNN pretrained on ChEMBL SMILES
- Agent: Initialized from prior, focused via RL/CL
- No pretrained model weights are publicly released
Evaluation
| Metric | Description | Notes |
|---|---|---|
| Docking score (Glide SP) | Predicted binding affinity (kcal/mol) | Lower is better; reference ligand: -10.907 |
| QED | Quantitative Estimate of Druglikeness | Range [0, 1] |
| Unique Bemis-Murcko scaffolds | Scaffold diversity measure | Averaged over triplicates |
| Cross-Tanimoto similarity | Intra-set compound diversity | Calculated on pooled triplicates |
| Tanimoto/ROCS similarity | Curriculum Objective metrics | 2D fingerprint and 3D shape similarity |
Hardware
- GPU: NVIDIA Tesla V100 (32 GB)
- Docking: AWS p3.8xlarge instance
- LigPrep parallelized over 8 CPU cores
- Glide docking parallelized over 48 CPU cores via DockStream
Artifacts
| Artifact | Type | License | Notes |
|---|---|---|---|
| REINVENT | Code | Apache-2.0 | De novo molecular design platform |
| CL Tutorial Notebook | Code | MIT | Jupyter notebook tutorial for curriculum learning |
Paper Information
Citation: Guo, J., Fialková, V., Arango, J. D., Margreitter, C., Janet, J. P., Papadopoulos, K., Engkvist, O., & Patronov, A. (2022). Improving de novo molecular design with curriculum learning. Nature Machine Intelligence, 4, 555-563. https://doi.org/10.1038/s42256-022-00494-4
@article{guo2022curriculum,
title={Improving de novo molecular design with curriculum learning},
author={Guo, Jeff and Fialkov{\'a}, Vendy and Arango, Juan Diego and Margreitter, Christian and Janet, Jon Paul and Papadopoulos, Kostas and Engkvist, Ola and Patronov, Atanas},
journal={Nature Machine Intelligence},
volume={4},
number={6},
pages={555--563},
year={2022},
publisher={Springer Nature},
doi={10.1038/s42256-022-00494-4}
}