Inceptionism Applied to Molecular Inverse Design
This is a Method paper that introduces PASITHEA, a gradient-based approach to de-novo molecular design inspired by inceptionism (deep dreaming) techniques from computer vision. The core contribution is a direct optimization framework that modifies molecular structures by backpropagating through a trained property-prediction network, with the molecular input (rather than weights) serving as the optimizable variable. PASITHEA is enabled by SELFIES, a surjective molecular string representation that guarantees 100% validity of generated molecules.
The Need for Direct Gradient-Based Molecular Optimization
Existing inverse molecular design methods, including variational autoencoders (VAEs), generative adversarial networks (GANs), reinforcement learning (RL), and genetic algorithms (GAs), share a common characteristic: they optimize molecules indirectly. VAEs and GANs learn distributions and scan latent spaces. RL agents learn policies from environmental rewards. GAs iteratively apply mutations and selections. None of these approaches directly maximize an objective function in a gradient-based manner with respect to the molecular representation itself.
This indirection has several consequences. VAE-based methods require learning a latent space, and the optimization happens in that space rather than directly on molecular structures. RL and GA methods require expensive function evaluations for each candidate molecule. The authors identify an opportunity to exploit gradients more directly by reversing the learning process of a neural network trained to predict molecular properties, thereby sidestepping latent spaces, policies, and population-based search entirely.
A second motivation is interpretability. By operating directly on the molecular representation (rather than a learned latent space), PASITHEA can reveal what a regression network has learned about structure-property relationships, a capability the authors frame as analogous to how deep dreaming reveals what image classifiers have learned about visual features.
Core Innovation: Inverting Regression Networks on SELFIES
PASITHEA’s key insight is a two-phase training procedure that repurposes the standard neural network training loop for molecule generation.
Phase 1: Prediction training. A fully connected neural network is trained to predict a real-valued chemical property (logP) from one-hot encoded SELFIES strings. The standard feedforward and backpropagation process updates the network weights to minimize mean squared error between predicted and ground-truth property values:
$$ \min_{\theta} \frac{1}{N} \sum_{i=1}^{N} (f_{\theta}(\mathbf{x}_i) - y_i)^2 $$
where $f_{\theta}$ is the neural network with parameters $\theta$, $\mathbf{x}_i$ is the one-hot encoded SELFIES input, and $y_i$ is the target logP value.
Phase 2: Inverse training (deep dreaming). The network weights $\theta$ are frozen. For a given input molecule $\mathbf{x}$ and a desired target property value $y_{\text{target}}$, the gradients are computed with respect to the input representation rather than the weights:
$$ \mathbf{x} \leftarrow \mathbf{x} - \eta \nabla_{\mathbf{x}} \mathcal{L}(f_{\theta}(\mathbf{x}), y_{\text{target}}) $$
This gradient descent on the input incrementally modifies the one-hot encoding of the molecular string, transforming it toward a structure whose predicted property matches the target value. At each step, the argmax function converts the continuous one-hot encoding back to a discrete SELFIES string, which always maps to a valid molecular graph due to the surjective property of SELFIES.
The role of SELFIES. The surjective mapping from strings to molecular graphs is essential. With SMILES, intermediate strings during optimization can become syntactically invalid (e.g., an unclosed ring like “CCCC1CCCCC”), producing no valid molecule. SELFIES enforces constraints that guarantee every string maps to a valid molecular graph, making the continuous gradient-based optimization feasible.
Input noise injection. Because inverse training transforms a one-hot encoding from binary values to real numbers, the discrete-to-continuous transition can cause convergence problems. The authors address this by initializing the input with noise: every zero in the one-hot encoding is replaced by a random number in $[0, k]$, where $k$ is a hyperparameter between 0.5 and 0.95. This smooths the optimization landscape and enables incremental molecular modifications rather than abrupt changes.
Experimental Setup on QM9 with LogP Optimization
Dataset and Property
The experiments use a random subset of 10,000 molecules from the QM9 dataset. The target property is the logarithm of the partition coefficient (logP), computed using RDKit. LogP measures lipophilicity, an important drug-likeness indicator that follows an approximately normal distribution in QM9 and has a nearly continuous range, making it suitable for gradient-based optimization.
Network Architecture
PASITHEA uses a fully connected neural network with four layers, each containing 500 nodes with ReLU activation. The loss function is mean squared error. Data is split 85%/15% for training/testing. The prediction model trains for approximately 1,500 epochs with an Adam optimizer and a learning rate of $1 \times 10^{-6}$.
For inverse training, the authors select a noise upper-bound of 0.9 and a learning rate of 0.01, chosen from hyperparameter tuning experiments that evaluate the percentage of molecules optimized toward the target property.
Optimization Targets
Two extreme logP targets are used: $+6$ (high lipophilicity) and $-6$ (low lipophilicity). These values exceed the range of logP values in the QM9 dataset (minimum: $-2.19$, maximum: $3.08$), testing whether the model can extrapolate beyond the training distribution.
Distribution Shifts and Interpretable Molecular Transformations
Distribution-Level Results
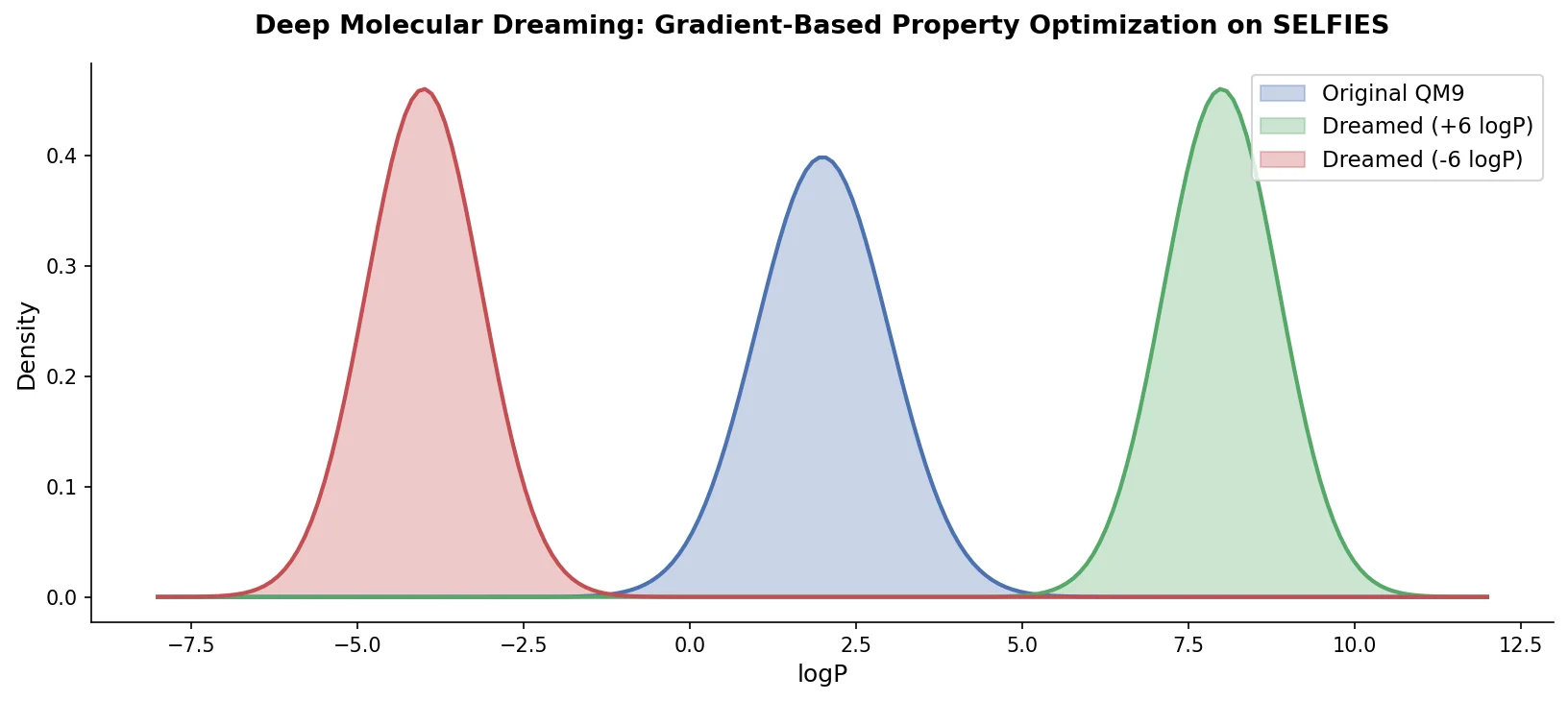
Applying deep dreaming to the full set of 10,000 molecules produces a clear shift in the logP distribution:
| Statistic | QM9 Original | Optimized (target +6) | Optimized (target -6) |
|---|---|---|---|
| Mean logP | 0.3909 | 1.8172 | -0.3360 |
| Min logP | -2.1903 | -0.8240 | -2.452 |
| Max logP | 3.0786 | 4.2442 | 0.9018 |
The optimized distributions extend beyond the original dataset’s property range. The right-shifted distribution (target +6) produces molecules with logP values up to 4.24, exceeding the original maximum of 3.08. The left-shifted distribution (target -6) reaches -2.45, below the original minimum. This indicates that PASITHEA can generate molecules with properties outside the training data bounds.
Additionally, 97.2% of the generated molecules do not exist in the original training set, indicating that the network is not memorizing data but rather using structural features to guide optimization. Some generated molecules contain more heavy atoms than the QM9 maximum of 9, since the SELFIES string length allows for larger structures.
Molecule-Level Interpretability
The stepwise molecular transformations reveal interpretable “strategies” the network employs:
Nitrogen appendage: When optimizing for lower logP, the network repeatedly appends nitrogen atoms to the molecule. The authors observe this as a consistent pattern across multiple test molecules, reflecting the known relationship between nitrogen content and reduced lipophilicity.
Length modulation: When optimizing for higher logP, the network tends to increase molecular chain length (e.g., extending a carbon chain). When optimizing for lower logP, it shortens chains. This captures the intuition that larger, more carbon-heavy molecules tend to be more lipophilic.
Bond order changes: The network replaces single bonds with double or triple bonds during optimization, demonstrating an understanding of the relationship between bonding patterns and logP.
Consistency across trials: Because the input initialization includes random noise, repeated trials with the same molecule produce different transformation sequences. Despite this stochasticity, the network applies consistent strategies across trials (e.g., always shortening chains for negative optimization), validating that it has learned genuine structure-property relationships.
Thermodynamic Stability
The authors assess synthesizability by computing heats of formation using MOPAC2016 at the PM7 level of theory. Some optimization trajectories move toward thermodynamically stable molecules (negative heats of formation), while others produce less stable structures. The authors acknowledge this limitation and propose multi-objective optimization incorporating stability as a future direction.
Comparison to VAEs
The key distinction from VAEs is where gradient computation occurs. In VAEs, a latent space is learned through encoding and decoding, and property optimization happens in that latent space. In PASITHEA, gradients are computed directly with respect to the molecular representation (SELFIES one-hot encoding). The authors argue this makes the approach more interpretable, since we can probe what the network learned about molecular structure without the “detour” through a latent space.
Limitations
The authors are forthright about the preliminary nature of these results:
- The method is demonstrated only on a small subset of QM9 with a single, computationally inexpensive property (logP).
- The simple four-layer architecture may not scale to larger molecular spaces or more complex properties.
- Generated molecules are not always thermodynamically stable, requiring additional optimization objectives.
- The approach has not been benchmarked against established methods (VAEs, GANs, RL) on standard generative benchmarks.
Reproducibility Details
Data
| Purpose | Dataset | Size | Notes |
|---|---|---|---|
| Training/Evaluation | QM9 (random subset) | 10,000 molecules | logP values computed via RDKit |
Algorithms
- Prediction training: 4-layer fully connected NN, 500 nodes/layer, ReLU activation, MSE loss, Adam optimizer, LR $1 \times 10^{-6}$, ~1,500 epochs, 85/15 train/test split
- Inverse training: Frozen weights, Adam optimizer, LR 0.01, noise upper-bound 0.9, logP targets of +6 and -6
- Heats of formation: MOPAC2016, PM7 level, geometry optimization with eigenvector following (EF)
Models
The architecture is a simple 4-layer MLP. No pre-trained weights are distributed, but the full code is available.
Evaluation
| Metric | Value | Notes |
|---|---|---|
| Novel molecules | 97.2% | Generated molecules not in training set |
| Max logP (target +6) | 4.2442 | Exceeds QM9 max of 3.0786 |
| Min logP (target -6) | -2.452 | Below QM9 min of -2.1903 |
Hardware
Not specified in the paper.
Artifacts
| Artifact | Type | License | Notes |
|---|---|---|---|
| Pasithea | Code | MIT | Official implementation |
Paper Information
Citation: Shen, C., Krenn, M., Eppel, S., & Aspuru-Guzik, A. (2021). Deep molecular dreaming: inverse machine learning for de-novo molecular design and interpretability with surjective representations. Machine Learning: Science and Technology, 2(3), 03LT02. https://doi.org/10.1088/2632-2153/ac09d6
@article{shen2021deep,
title={Deep molecular dreaming: inverse machine learning for de-novo molecular design and interpretability with surjective representations},
author={Shen, Cynthia and Krenn, Mario and Eppel, Sagi and Aspuru-Guzik, Al{\'a}n},
journal={Machine Learning: Science and Technology},
volume={2},
number={3},
pages={03LT02},
year={2021},
publisher={IOP Publishing},
doi={10.1088/2632-2153/ac09d6}
}