A Controlled Generation Framework for Target-Specific Drug Design
This is a Method paper that introduces CogMol (Controlled Generation of Molecules), an end-to-end framework for de novo drug design. The primary contribution is a pipeline that combines a SMILES-based Variational Autoencoder (VAE) with multi-attribute controlled latent space sampling (CLaSS) to generate novel drug-like molecules with high binding affinity to specified protein targets, off-target selectivity, and favorable drug-likeness properties. The framework operates on protein sequence embeddings, allowing it to generalize to unseen target proteins without model retraining.
Multi-Constraint Drug Design for Novel Viral Targets
Traditional drug discovery costs 2-3 billion USD and takes over a decade with less than 10% success rate. Generating drug molecules requires satisfying multiple competing objectives simultaneously: target binding affinity, off-target selectivity, synthetic accessibility, drug-likeness, and low toxicity. Prior generative approaches using reinforcement learning or Bayesian optimization are computationally expensive and typically require fine-tuning on target-specific ligand libraries, making them unable to generalize to unseen protein targets.
The emergence of SARS-CoV-2 in 2020 created an urgent need for antiviral drug candidates targeting novel viral proteins. Because no binding affinity data existed for these new targets, and the viral proteins were not closely related to proteins in existing databases like BindingDB, existing target-specific generative frameworks could not be directly applied. CogMol addresses this by using pre-trained protein sequence embeddings from UniRep (trained on 24 million UniRef50 sequences) rather than learning protein representations from the limited BindingDB training set.
Controlled Latent Space Sampling with Pre-trained Protein Embeddings
CogMol’s core innovation is a three-component architecture that enables multi-constraint molecule generation for unseen targets:
1. SMILES VAE with adaptive pre-training. A Variational Autoencoder is first trained unsupervised on the MOSES/ZINC dataset (1.6M molecules), then jointly fine-tuned with QED and SA property predictors on BindingDB molecules. The standard VAE objective is:
$$\mathcal{L}_{\text{VAE}}(\theta, \phi) = \mathbb{E}_{p(x)} \left\{ \mathbb{E}_{q_\phi(z|x)} [\log p_\theta(x|z)] - D_{\text{KL}}(q_\phi(z|x) | p(z)) \right\}$$
where $q_\phi(z|x) = \mathcal{N}(z; \mu(x), \Sigma(x))$ specifies a diagonal Gaussian encoder distribution.
2. Protein-molecule binding affinity predictor. A regression model takes pre-trained UniRep protein sequence embeddings and molecule latent embeddings $z$ as input and predicts pIC50 binding affinity ($= -\log(\text{IC50})$). Because UniRep embeddings capture sequence, structural, and functional relationships from a large unsupervised corpus, the predictor can estimate binding affinity for novel target sequences not present in the training data.
3. CLaSS controlled sampling. The Conditional Latent attribute Space Sampling scheme generates molecules satisfying multiple constraints (affinity, QED, selectivity) through rejection sampling in the VAE latent space:
$$p(\mathbf{x} | \mathbf{a}) = \mathbb{E}_{\mathbf{z}} [p(\mathbf{z} | \mathbf{a}) , p(\mathbf{x} | \mathbf{z})] \approx \mathbb{E}_{\mathbf{z}} [\hat{p}_\xi(\mathbf{z} | \mathbf{a}) , p_\theta(\mathbf{x} | \mathbf{z})]$$
where $\mathbf{a} = [a_1, a_2, \ldots, a_n]$ is a set of independent attribute constraints. The conditional density $\hat{p}_\xi(\mathbf{z} | \mathbf{a})$ is approximated using a Gaussian mixture model $Q_\xi(\mathbf{z})$ and per-attribute classifiers $q_\xi(a_i | \mathbf{z})$, with Bayes’ rule and conditional independence assumptions. The acceptance probability equals the product of all attribute predictor scores, enabling efficient multi-constraint sampling without surrogate model or policy learning.
Selectivity modeling. Off-target selectivity for a molecule $m$ against target $T$ is defined as:
$$\text{Sel}_{T,m} = \text{BA}(T, m) - \frac{1}{k} \sum_{i=1}^{k} \text{BA}(T_i, m)$$
where $\text{BA}(T, m)$ is binding affinity to the target and $T_i$ are $k$ randomly selected off-targets. This selectivity score is incorporated as a control attribute during CLaSS sampling.
Experimental Setup: COVID-19 Targets and In Silico Screening
Target proteins. CogMol was applied to three SARS-CoV-2 targets not present in BindingDB: NSP9 Replicase dimer, Main Protease (Mpro), and the Receptor-Binding Domain (RBD) of the spike protein. A cancer target (human HDAC1) with low ligand coverage in the training data was also evaluated.
Training data. The SMILES VAE was trained on the MOSES benchmark (1.6M molecules from ZINC). The binding affinity predictor used curated IC50 data from BindingDB as reported in DeepAffinity, with all protein classes included in training.
CLaSS controlled generation. Molecules were generated with simultaneous constraints on binding affinity (> 0.5 normalized), QED (> 0.8 normalized), and selectivity (> 0.5 normalized). Approximately 1000 molecules per target were selected for downstream evaluation.
In silico screening pipeline. Generated molecules underwent:
- Toxicity prediction via a multi-task deep neural network (MT-DNN) on 12 Tox21 in vitro endpoints and ClinTox clinical trial failure
- Binding affinity rescoring with a higher-accuracy SMILES-level predictor
- Blind docking (5 independent runs per molecule) using AutoDock Vina against target protein structures
- Synthetic feasibility assessment using a retrosynthetic algorithm based on the Molecular Transformer trained on patent reaction data
Baselines. VAE performance was benchmarked against models from the MOSES platform. CLaSS-accepted molecules were compared against randomly sampled molecules from the latent space. Generated molecules were compared against FDA-approved drugs for toxicity and synthesizability.
Key Results
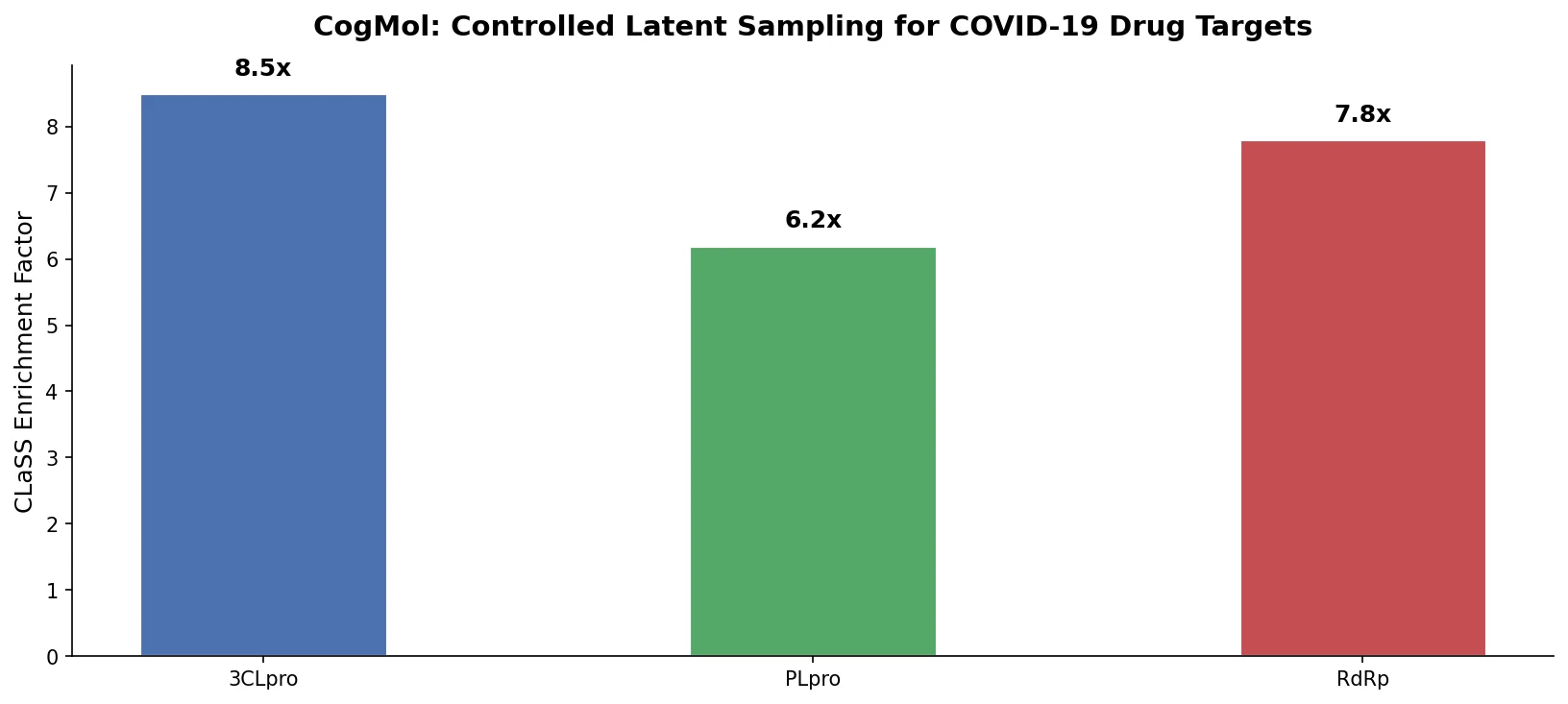
CLaSS enrichment (Table 1). CLaSS consistently produced higher fractions of molecules meeting all criteria compared to random sampling. For the triple constraint (affinity > 0.5, QED > 0.8, selectivity > 0.5), the enrichment was substantial: 6.9% vs. 0.7% for NSP9, 9.0% vs. 0.9% for RBD, and 10.4% vs. 1.1% for Mpro.
| Target | CLaSS (Aff+QED+Sel) | Random (Aff+QED+Sel) | Enrichment |
|---|---|---|---|
| NSP9 | 6.9% | 0.7% | ~10x |
| RBD | 9.0% | 0.9% | ~10x |
| Mpro | 10.4% | 1.1% | ~9.5x |
Docking results (Table 3). 87-95% of high-affinity generated molecules showed docking binding free energy (BFE) below -6 kcal/mol, with minimum BFEs reaching -8.6 to -9.5 kcal/mol depending on the target.
Novelty. The likelihood of generating an exact duplicate of a training molecule was 2% or less. Against the full PubChem database (~103M molecules), exact matches ranged from 3.7% to 9.5%. Generated molecules also showed novel chemical scaffolds as confirmed by high Frechet ChemNet Distance.
Synthesizability. Generated molecules for COVID-19 targets showed 85-90% synthetic feasibility using retrosynthetic analysis, exceeding the ~78% rate of FDA-approved drugs.
Toxicity. Approximately 70% of generated parent molecules and ~80% of predicted metabolites were toxic in 0-1 endpoints out of 13, comparable to FDA-approved drugs.
Generated Molecules Show Favorable Binding and Drug-Like Properties
CogMol demonstrates that controlled latent space sampling with pre-trained protein embeddings can generate novel, drug-like molecules for unseen viral targets. The key findings are:
- CLaSS provides roughly 10x enrichment over random latent space sampling for molecules satisfying all three constraints (affinity, QED, selectivity).
- Generated molecules bind favorably to druggable pockets in target protein 3D structures, even though the generation model uses only 1D sequence information.
- Some generated SMILES matched existing PubChem molecules with known biological activity, suggesting the model identifies chemically relevant regions of molecular space.
- The framework generalizes across targets of varying novelty, with Mpro (more similar to training proteins) yielding easier generation than NSP9 or RBD.
Limitations. The authors note that no wet-lab validation was performed on generated candidates. There may be divergence between ML-predicted properties and experimental measurements. The binding affinity predictor’s accuracy is bounded by the quality and coverage of BindingDB training data. Selectivity modeling uses a random sample of off-targets rather than a pharmacologically curated panel.
Future directions. The authors propose incorporating additional contexts beyond target protein (e.g., metabolic pathways), adding more pharmacologically relevant controls, and weighting objectives by relative importance.
Reproducibility Details
Data
| Purpose | Dataset | Size | Notes |
|---|---|---|---|
| VAE pre-training | MOSES/ZINC | 1.6M train, 176K test | Publicly available benchmark |
| VAE adaptive training | BindingDB (DeepAffinity split) | ~27K protein-ligand pairs | Curated IC50 data |
| Protein embeddings | UniRef50 via UniRep | 24M sequences | Pre-trained, publicly available |
| Toxicity prediction | Tox21 + ClinTox | 12 in vitro + clinical endpoints | Public benchmark datasets |
| Docking validation | PDB structures | 3 SARS-CoV-2 targets | Public crystal structures |
Algorithms
- VAE architecture: SMILES encoder-decoder with diagonal Gaussian latent space, jointly trained with QED and SA regressors
- CLaSS: rejection sampling from Gaussian mixture model of latent space with per-attribute classifiers
- Binding affinity: regression on concatenated UniRep protein embeddings and VAE molecule embeddings
- Selectivity: excess binding affinity over average of $k$ random off-targets
Models
- SMILES VAE with adaptive pre-training (ZINC then BindingDB)
- Multi-task toxicity classifier (MT-DNN) for Tox21 and ClinTox endpoints
- Binding affinity predictor (latent-level for generation, SMILES-level for screening)
- Retrosynthetic predictor based on Molecular Transformer
Evaluation
| Metric | Value | Baseline | Notes |
|---|---|---|---|
| Validity | 90% | - | Generated SMILES |
| Uniqueness | 99% | - | Among valid molecules |
| Filter pass | 95% | - | Relevant chemical filters |
| Docking BFE < -6 kcal/mol | 87-95% | - | Varies by target |
| Synthetic feasibility | 85-90% | 78% (FDA drugs) | COVID-19 targets |
| Low toxicity (0-1 endpoints) | ~70% parent, ~80% metabolite | Comparable to FDA drugs | MT-DNN prediction |
Hardware
The paper does not specify GPU types or training times. The work was funded internally by IBM Research.
Artifacts
| Artifact | Type | License | Notes |
|---|---|---|---|
| CogMol (GitHub) | Code | Apache-2.0 | Official implementation |
| ~3500 generated molecules | Dataset | Open license | For three SARS-CoV-2 targets |
Paper Information
Citation: Chenthamarakshan, V., Das, P., Hoffman, S. C., Strobelt, H., Padhi, I., Lim, K. W., Hoover, B., Manica, M., Born, J., Laino, T., & Mojsilovic, A. (2020). CogMol: Target-Specific and Selective Drug Design for COVID-19 Using Deep Generative Models. Advances in Neural Information Processing Systems, 33, 4320-4332.
@inproceedings{chenthamarakshan2020cogmol,
title={CogMol: Target-Specific and Selective Drug Design for COVID-19 Using Deep Generative Models},
author={Chenthamarakshan, Vijil and Das, Payel and Hoffman, Samuel C. and Strobelt, Hendrik and Padhi, Inkit and Lim, Kar Wai and Hoover, Benjamin and Manica, Matteo and Born, Jannis and Laino, Teodoro and Mojsilovi{\'c}, Aleksandra},
booktitle={Advances in Neural Information Processing Systems},
volume={33},
pages={4320--4332},
year={2020}
}