Reinterpreting Goal-Directed Generation Failures as QSAR Model Issues
This is an Empirical study that challenges a widely cited finding about failure modes in goal-directed molecular generation. Renz et al. (2019) had shown that when molecules are optimized against a machine learning scoring function, control models trained on the same data distribution assign much lower scores to the generated molecules. This was interpreted as evidence that generation algorithms exploit model-specific biases. Langevin et al. demonstrate that this divergence is already present in the original data distribution and is attributable to disagreement among the QSAR classifiers, not to flaws in the generation algorithms themselves.
Why QSAR Model Agreement Matters for Molecular Generation
Goal-directed generation uses a scoring function (typically a QSAR model) to guide the design of molecules that maximize predicted activity. In the experimental framework from Renz et al., three Random Forest classifiers are trained: an optimization model $C_{opt}$ on Split 1, a model control $C_{mc}$ on Split 1 with a different random seed, and a data control $C_{dc}$ on Split 2. Each returns a confidence score ($S_{opt}$, $S_{mc}$, $S_{dc}$). The expectation is that molecules with high $S_{opt}$ should also score highly under $S_{mc}$ and $S_{dc}$, since all three models are trained on the same data distribution for the same target.
Renz et al. observed that during optimization, $S_{mc}$ and $S_{dc}$ diverge from $S_{opt}$, reaching substantially lower values. This was interpreted as goal-directed generation exploiting biases unique to the optimization model. The recommendation was to halt generation when control scores stop increasing, requiring a held-out dataset for a control model, which may not be feasible in low-data regimes.
The key insight of Langevin et al. is that nobody had checked whether this score disagreement existed before generation even began. If the classifiers already disagree on high-scoring molecules in the original dataset, the divergence during generation is expected behavior, not evidence of algorithmic failure.
Pre-Existing Classifier Disagreement Explains the Divergence
The core contribution is showing that the gap between optimization and control scores is a property of the QSAR models, not of the generation algorithms.
The authors introduce a held-out test set (10% of the data, used for neither training split) and augment it via Topliss tree enumeration to produce structural analogs for smoother statistical estimates. On this held-out set, they compute the Mean Average Difference (MAD) between $S_{opt}$ and control scores as a function of $S_{opt}$:
$$ \text{MAD}(x) = \frac{1}{|\{i : S_{opt}(x_i) \geq x\}|} \sum_{S_{opt}(x_i) \geq x} |S_{opt}(x_i) - S_{dc}(x_i)| $$
On the three original datasets (DRD2, EGFR, JAK2), the MAD between $S_{opt}$ and $S_{dc}$ grows substantially with $S_{opt}$, reaching approximately 0.3 for the highest-scoring molecules. For EGFR, even the top molecules (with $S_{opt}$ between 0.5 and 0.6) have $S_{dc}$ below 0.2. This disagreement exists entirely within the original data distribution, before any generative algorithm is applied.
The authors formalize this with tolerance intervals. At each generation time step $t$, the distribution of optimization scores is $P_t[S_{opt}(x)]$. From the held-out set, the conditional distributions $P[S_{dc}(x) | S_{opt}(x)]$ and $P[S_{mc}(x) | S_{opt}(x)]$ are estimated empirically. The expected control scores at time $t$ are then:
$$ \mathbb{E}[S_{dc}] = \int P[S_{dc}(x) | S_{opt}(x)] \cdot P_t[S_{opt}(x)] , dS_{opt} $$
By sampling from these distributions, the authors construct 95% tolerance intervals for the expected control scores at each time step. The observed trajectories of $S_{mc}$ and $S_{dc}$ during generation fall within these intervals, demonstrating that the divergence is fully explained by pre-existing classifier disagreement.
Experimental Setup: Original Reproduction and Corrected Experiments
Reproduction of Renz et al.
The original experimental framework uses three datasets from ChEMBL: DRD2 (842 molecules, 59 actives), EGFR (842 molecules, 40 actives), and JAK2 (667 molecules, 140 actives). These are small, noisy, and chemically diverse. Three goal-directed generation algorithms are tested:
| Algorithm | Type | Mechanism |
|---|---|---|
| Graph GA | Genetic algorithm on molecular graphs | Mutation and crossover of molecular graphs |
| SMILES-LSTM | Recurrent neural network | Hill-climbing fine-tuning on best molecules |
| MSO | Particle swarm in CDDD latent space | Multiple swarm optimization |
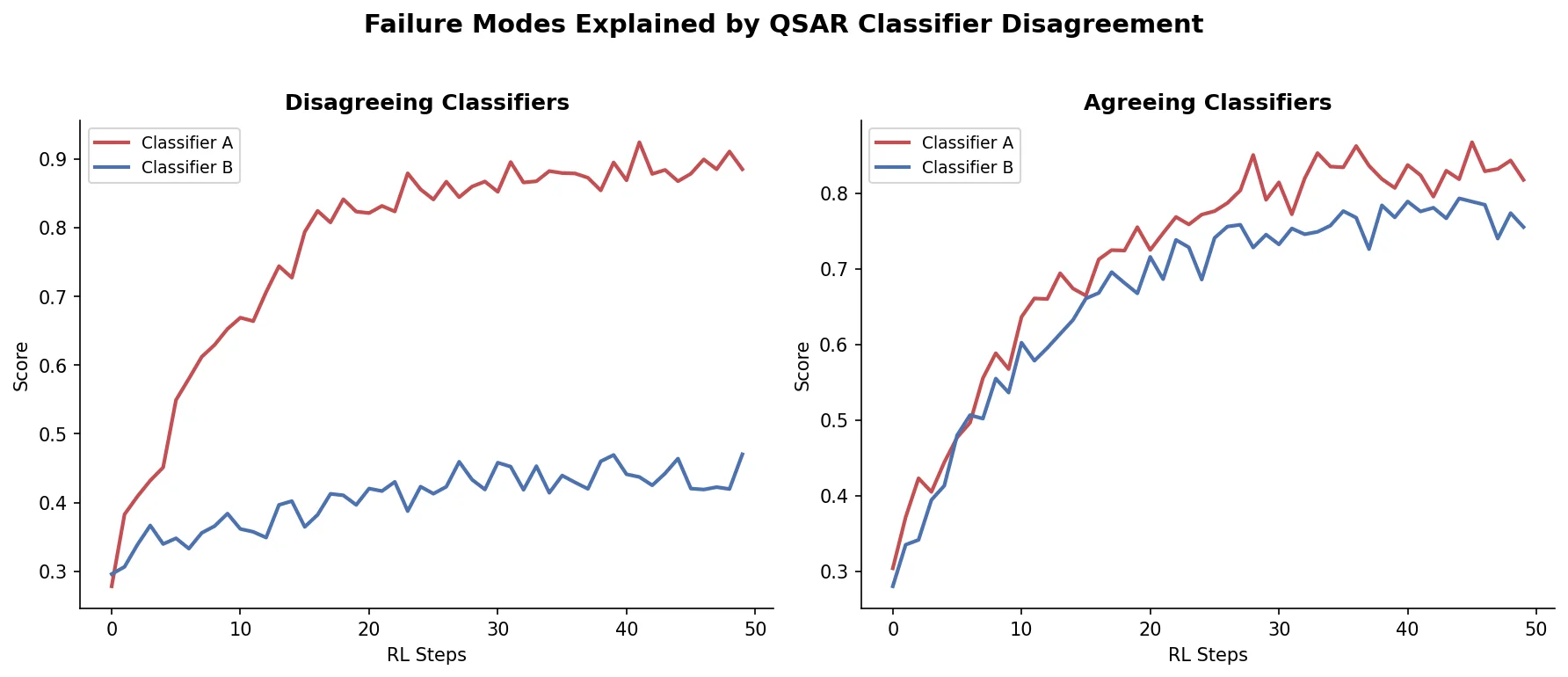
All algorithms are run for 151 epochs with 10 runs each. The reproduction confirms the findings of Renz et al.: $S_{mc}$ and $S_{dc}$ diverge from $S_{opt}$ during optimization.
Tolerance interval analysis
The held-out set is augmented using Topliss tree enumeration on phenyl rings, providing structural analogs that are reasonable from a medicinal chemistry perspective. The optimization score range is divided into 25 equal bins, and for each molecule at each time step, 10 samples from the conditional control score distribution are drawn to construct empirical tolerance intervals.
Corrected experiments with adequate models
To test whether generation algorithms actually exploit biases when the classifiers agree, the authors construct two tasks where optimization and control models correlate well:
- ALDH1 dataset: 464 molecules from LIT-PCBA, split using similarity-based pairing to maximize intra-pair chemical similarity. This ensures both splits sample similar chemistry.
- Modified JAK2: The same JAK2 dataset but with Random Forest hyperparameters adjusted (200 trees instead of 100, minimum 3 samples per leaf instead of 1) to reduce overfitting to spurious correlations.
In both cases, $S_{opt}$, $S_{mc}$, and $S_{dc}$ agree well on the held-out test set. The starting population for generation is set to the held-out test set (rather than random ChEMBL molecules) to avoid building in a distribution shift.
Findings: No Algorithmic Failure When Models Agree
On the corrected experimental setups (ALDH1 and modified JAK2), there is no major divergence between optimization and control scores during generation. The three algorithms produce molecules that score similarly under all three classifiers.
Key findings:
Pre-existing disagreement explains divergence: On all three original datasets, the divergence between $S_{opt}$ and control scores during generation falls within the tolerance intervals predicted from the initial data distribution alone. The generation algorithms are not exploiting model-specific biases beyond what already exists in the data.
Split similarity bias is also pre-existing: Renz et al. observed that generated molecules are more similar to Split 1 (used to train $C_{opt}$) than Split 2. The authors show this bias is already present in the top-5 percentile of the held-out set: on EGFR and DRD2, high-scoring molecules are inherently more similar to Split 1.
Appropriate model design resolves the issue: When Random Forest hyperparameters are chosen to avoid overfitting (more trees, higher minimum samples per leaf), or when data splits are constructed to be chemically balanced, the classifiers agree and the generation algorithms behave as expected.
Quality problems remain independent: Even when optimization and control scores align, the generated molecules can still be poor drug candidates (unreactive, unsynthesizable, containing unusual fragments). The score divergence issue and the chemical quality issue are separate problems.
Limitations acknowledged by the authors
- The study focuses on Random Forest classifiers with ECFP fingerprints. The behavior of other model types (e.g., graph neural networks) and descriptor types is not fully explored, though supplementary results show similar patterns with physico-chemical descriptors and Atom-Pair fingerprints.
- The corrected ALDH1 task uses a relatively small dataset (464 molecules) with careful split construction. Scaling this approach to larger, more heterogeneous datasets is not demonstrated.
- The authors note that their results do not prove generation algorithms never exploit biases; they show that the specific evidence from Renz et al. can be explained without invoking algorithmic failure.
- The problem of low-quality generated molecules (poor synthesizability, unusual fragments) remains unresolved and is acknowledged as an open question.
Reproducibility Details
Data
| Purpose | Dataset | Size | Notes |
|---|---|---|---|
| Original tasks | DRD2, EGFR, JAK2 | 842, 842, 667 molecules | Extracted from ChEMBL; small with few actives |
| New task | ALDH1 | 464 molecules (173 with purine substructure) | Extracted from LIT-PCBA; similarity-based split |
| Augmentation | Topliss tree analogs | ~10x augmentation of held-out set | Structural analogs via phenyl ring enumeration |
Algorithms
Three goal-directed generation algorithms from the original Renz et al. study:
- Graph GA: Genetic algorithm on molecular graphs (Jensen, 2019)
- SMILES-LSTM: Hill-climbing on LSTM-generated SMILES (Segler et al., 2018)
- MSO: Multi-Swarm Optimization in CDDD latent space (Winter et al., 2019)
All run for 151 epochs, 10 runs each.
Models
Random Forest classifiers (scikit-learn) with:
- ECFP fingerprints (radius 2, 1024 bits, RDKit)
- Default parameters for original tasks
- Modified parameters for JAK2 correction: 200 trees, min 3 samples per leaf
Evaluation
| Metric | Purpose | Notes |
|---|---|---|
| Mean Average Difference (MAD) | Measures disagreement between optimization and control scores | Computed as function of $S_{opt}$ on held-out set |
| 95% tolerance intervals | Expected range of control scores given optimization scores | Empirical, constructed from held-out set |
| Tanimoto similarity | Split bias assessment | Morgan fingerprints, radius 2, 1024 bits |
| ROC-AUC | Classifier predictive performance | Used to verify models have comparable accuracy |
Hardware
Not specified in the paper.
Artifacts
| Artifact | Type | License | Notes |
|---|---|---|---|
| Code and datasets | Code | Apache-2.0 | Fork of Renz et al. codebase with modifications |
Paper Information
Citation: Langevin, M., Vuilleumier, R., & Bianciotto, M. (2022). Explaining and avoiding failure modes in goal-directed generation of small molecules. Journal of Cheminformatics, 14, 20. https://doi.org/10.1186/s13321-022-00601-y
@article{langevin2022explaining,
title={Explaining and avoiding failure modes in goal-directed generation of small molecules},
author={Langevin, Maxime and Vuilleumier, Rodolphe and Bianciotto, Marc},
journal={Journal of Cheminformatics},
volume={14},
number={1},
pages={20},
year={2022},
publisher={Springer},
doi={10.1186/s13321-022-00601-y}
}