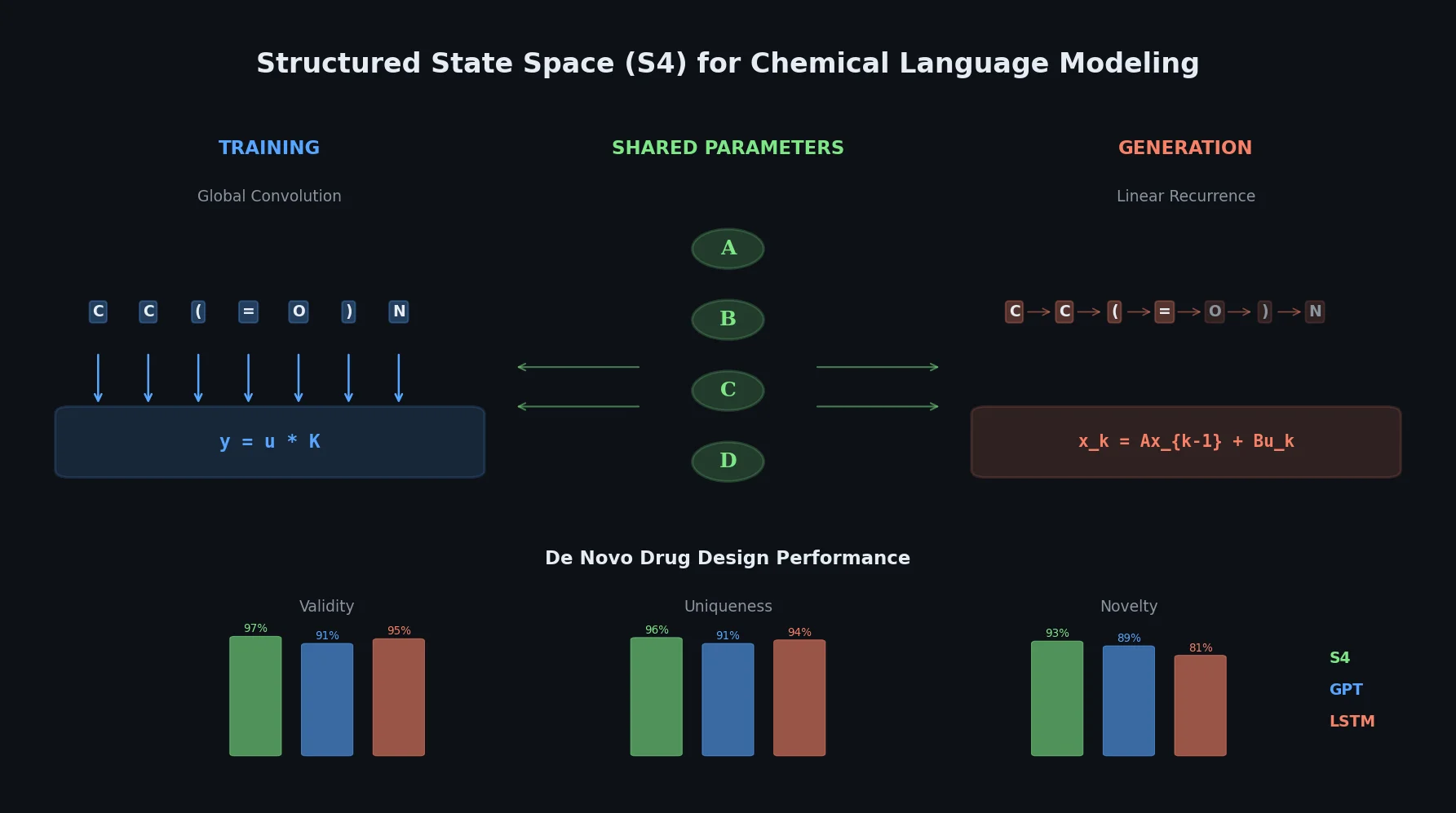
Structured State Spaces Meet Chemical Language Modeling
This is a Method paper that introduces structured state space sequence (S4) models to chemical language modeling (CLM) for de novo drug design. S4 models have a dual formulation: they process entire input sequences via convolution during training (like Transformers) and generate sequences element-by-element via recurrence during inference (like LSTMs). The authors benchmark S4 against LSTM and GPT architectures across multiple drug discovery tasks, including drug-like molecule generation, bioactivity learning, chemical space exploration, natural product design, and prospective kinase inhibitor design validated by molecular dynamics simulations.
Bridging the LSTM-Transformer Gap in Molecular Generation
Chemical language models (CLMs) generate molecules by learning the “chemical language” of SMILES string representations. The two dominant architectures for CLMs are LSTMs and GPTs, each with complementary strengths and limitations:
- LSTMs generate sequences recurrently (element-by-element), which enables efficient generation and good learning of local/short-range dependencies. However, their sequential information bottleneck limits learning of global sequence properties.
- GPTs (Transformer decoders) process the entire input at once, better capturing global properties like bioactivity. However, they become increasingly compute-intensive for longer SMILES strings and struggle with chemical space exploration at higher sampling temperatures.
Complex molecular properties like bioactivity can emerge from separated portions of a SMILES string (e.g., distant functional groups in the linear notation). Neither architecture fully addresses the need to learn these long-range dependencies while maintaining efficient, robust generation. The chemical space, estimated at up to $10^{60}$ small molecules, demands models that can both capture complex property relationships and explore diverse scaffolds efficiently.
The Dual Nature of S4: Convolution Meets Recurrence
S4 models are built on discrete state space models, which map an input sequence $\mathbf{u}$ to an output sequence $\mathbf{y}$ through learnable parameters $\overline{\mathbf{A}} \in \mathbb{R}^{N \times N}$, $\overline{\mathbf{B}} \in \mathbb{R}^{N \times 1}$, $\overline{\mathbf{C}} \in \mathbb{R}^{1 \times N}$, and $\overline{\mathbf{D}} \in \mathbb{R}^{1 \times 1}$:
$$ x_{k} = \overline{\mathbf{A}} x_{k-1} + \overline{\mathbf{B}} u_{k} $$
$$ y_{k} = \overline{\mathbf{C}} x_{k} + \overline{\mathbf{D}} u_{k} $$
This linear recurrence can equivalently be “unrolled” into a global convolution:
$$ \mathbf{y} = \mathbf{u} * \overline{\mathbf{K}} $$
where $\overline{\mathbf{K}}$ is a convolution filter parameterized by $\overline{\mathbf{A}}$, $\overline{\mathbf{B}}$, and $\overline{\mathbf{C}}$. This duality is the core innovation for CLMs:
- Training: S4 uses the convolutional formulation to learn from entire SMILES sequences simultaneously, capturing global molecular properties.
- Generation: S4 switches to the recurrent formulation, producing SMILES tokens one at a time for efficient, robust chemical space exploration.
S4 addresses the numerical instabilities of naive state space models through high-order polynomial projection operators (HiPPO) and reduction to the stable Cauchy kernel computation, enabling effective learning of long-range dependencies.
For molecular ranking after fine-tuning, the log-likelihood score subtracts the pre-training likelihood to isolate target-specific information:
$$ \mathcal{L}_{\text{score}}(\mathbf{M}) = \mathcal{L}(\mathbf{M}_{\text{ft}}) - \mathcal{L}(\mathbf{M}_{\text{pt}}) $$
where $\mathcal{L}(\mathbf{M}_{\text{ft}})$ and $\mathcal{L}(\mathbf{M}_{\text{pt}})$ are the fine-tuned and pre-trained model log-likelihoods, respectively.
Benchmarking S4 Across Drug Discovery Tasks
Drug-like molecule generation
All three CLMs (S4, LSTM, GPT) were pre-trained on 1.9M canonical SMILES from ChEMBL v31 (molecules with fewer than 100 tokens). Each model generated 102,400 SMILES strings de novo.
| Model | Valid | Unique | Novel |
|---|---|---|---|
| S4 | 99,268 (97%) | 98,712 (96%) | 95,552 (93%) |
| LSTM | 97,151 (95%) | 96,618 (94%) | 82,988 (81%) |
| GPT | 93,580 (91%) | 93,263 (91%) | 91,590 (89%) |
S4 produces the most valid, unique, and novel molecules. Error analysis reveals that each architecture shows different failure modes: LSTMs struggle most with branching errors, GPTs with ring and bond assignment errors, while S4 generates fewer branching and ring errors but more bond assignment errors than LSTM. This pattern supports the hypothesis that S4 captures long-range dependencies (branching, ring opening/closure) better while local dependencies (bond assignment) are handled better by recurrent processing.
Bioactivity learning via transfer learning
Five fine-tuning campaigns were conducted on targets from the LIT-PCBA dataset: PKM2, MAPK1, GBA, mTORC1, and TP53. After fine-tuning, models ranked held-out test molecules by learned log-likelihoods to evaluate bioactive compound prioritization.
S4 outperformed both benchmarks across targets. Wilcoxon signed-rank tests on pooled scores confirmed statistically significant superiority:
- S4 vs. LSTM: $p$ [top 10] = 8.41e-6, $p$ [top 50] = 2.93e-7, $p$ [top 100] = 1.45e-7
- S4 vs. GPT: $p$ [top 10] = 2.33e-3, $p$ [top 50] = 3.72e-3, $p$ [top 100] = 2.61e-2
TP53 was the most challenging target, where no model consistently retrieved actives in the top 10, possibly due to activity cliffs in the test set.
Chemical space exploration with temperature sampling
Models were evaluated across sampling temperatures from $T = 1.0$ to $T = 2.0$ on three metrics: SMILES validity, rediscovery rate of known actives, and scaffold diversity. Key findings:
- Validity: S4 and LSTM maintain higher validity than GPT at elevated temperatures (GPT median validity drops below 40% at high T).
- Rediscovery: S4 outperforms LSTM in rediscovering bioactive molecules at all temperatures.
- Scaffold diversity: LSTM achieves the highest number of unique scaffold clusters (median 6,602 at $T = 1.75$), with S4 as close second (6,520 clusters).
S4 provides the best balance between bioactivity capture and structural diversity.
Natural product design
Models were trained on 32,360 large natural product SMILES (length > 100 tokens) from the COCONUT database and used to generate 102,400 designs each.
| Metric | S4 | LSTM | GPT | Training Set |
|---|---|---|---|---|
| Valid | 82,633 (81%) | 76,264 (74%) | 70,117 (68%) | n.a. |
| Unique | 53,293 (52%) | 51,326 (50%) | 50,487 (49%) | n.a. |
| Novel | 40,897 (40%) | 43,245 (42%) | 43,168 (42%) | n.a. |
| NP-likeness | 1.6 +/- 0.7 | 1.5 +/- 0.7 | 1.5 +/- 0.7 | 1.6 +/- 0.7 |
S4 designs the most valid molecules (6,000 to 12,000 more than benchmarks) and achieves significantly higher NP-likeness ($p = 1.41 \times 10^{-53}$ vs. LSTM, $p = 1.02 \times 10^{-82}$ vs. GPT). S4 also achieves the lowest Kolmogorov-Smirnov distances to the training/test distributions across multiple structural properties (sp3 carbons, aliphatic rings, spiro atoms, molecular weight, fused ring size, heavy atoms).
For computational efficiency, S4 trains as fast as GPT (both approximately 1.3x faster than LSTM) and generates fastest among all architectures.
Prospective MAPK1 inhibitor design
The pre-trained S4 model was fine-tuned on 68 manually curated MAPK1 inhibitors ($K_i < 1 \mu M$) from ChEMBL v33. The last five fine-tuning epochs generated 256K molecules across five temperature values. After ranking and filtering by log-likelihood score and scaffold similarity, the top 10 designs were evaluated via Umbrella Sampling molecular dynamics simulations.
Eight out of ten designs showed high predicted affinity, with $\Delta G$ values ranging from $-10.3 \pm 0.6$ to $-23 \pm 4$ kcal/mol. These affinities are comparable to or exceed those of the closest known active neighbors ($\Delta G = -9.1 \pm 0.8$ to $-13 \pm 2$ kcal/mol). The most potent predicted design (molecule 2, $\Delta G = -23 \pm 4$ kcal/mol) engages extensively with the MAPK1 binding pocket, though synthetic accessibility may be limited. Several designs incorporate halogen substitutions favorable for MAPK1 inhibition, consistent with known structure-activity relationships.
S4 Combines the Best of LSTMs and GPTs for Molecular Design
The main findings of this study are:
- S4 outperforms both LSTM and GPT in learning complex molecular properties like bioactivity, while maintaining competitive or superior performance in syntax learning and chemical space exploration.
- The dual formulation is key: holistic training (convolution) enables better capture of global molecular properties, while recurrent generation preserves robust chemical syntax and diverse scaffold exploration.
- S4 is especially strong for longer sequences: natural product design (SMILES > 100 tokens) shows the largest advantages over benchmarks in validity and property matching.
- Prospective validation: 8/10 S4-designed MAPK1 inhibitors are predicted as highly active by molecular dynamics, with affinities comparable to or exceeding known actives.
Limitations acknowledged by the authors:
- All evaluations are computational; no wet-lab experimental validation is reported.
- Bioactivity evaluation relies on likelihood-based ranking, which is an indirect proxy.
- The MD simulations, while more rigorous than simple docking, still represent in silico predictions.
- SMILES augmentation and improved ranking protocols could further boost performance.
Future directions include application to macrocyclic peptides and protein sequences, organic reaction planning, structure-based drug design, and integration with wet-lab experimental validation.
Reproducibility Details
Data
| Purpose | Dataset | Size | Notes |
|---|---|---|---|
| Pre-training | ChEMBL v31 | 1.9M SMILES | Molecules with SMILES length <= 100 tokens |
| Fine-tuning (bioactivity) | LIT-PCBA (5 targets) | 11-56 actives + ~10K inactives per target | PKM2, MAPK1, GBA, mTORC1, TP53 |
| Natural product training | COCONUT | 32,360 SMILES | SMILES length > 100 tokens |
| Prospective fine-tuning | ChEMBL v33 (MAPK1) | 68 inhibitors | $K_i < 1 \mu M$, target ID CHEMBL4040 |
Algorithms
- Pre-training: next-token prediction on SMILES strings
- Fine-tuning: transfer learning with early stopping (patience 5, tolerance $10^{-5}$)
- Molecule ranking: log-likelihood scoring with pre-training bias subtraction (Eq. 5)
- Temperature sampling: $T$ from 1.0 to 2.0 (step 0.25) for chemical space exploration
Models
- S4: Structured state space sequence model with HiPPO initialization; hyperparameter search over 242 + 108 configurations
- LSTM: 40 configurations optimized via random search
- GPT: 35 configurations optimized via random search
- All models share the same pre-training data and fine-tuning protocol for fair comparison
Evaluation
| Metric | Best Model | Value | Notes |
|---|---|---|---|
| Validity (ChEMBL) | S4 | 97% | Out of 102,400 generated SMILES |
| Uniqueness (ChEMBL) | S4 | 96% | Among valid designs |
| Novelty (ChEMBL) | S4 | 93% | Not in training set |
| Bioactivity ranking (top 10) | S4 | Significant (p = 8.41e-6 vs LSTM) | Wilcoxon signed-rank test |
| NP validity | S4 | 81% | COCONUT, SMILES > 100 tokens |
| MAPK1 inhibitor success | S4 | 8/10 designs active | Validated by MD (Umbrella Sampling) |
Hardware
- Hyperparameter search: NVIDIA A100 40GB GPUs
- LSTM/GPT search: 5 days on single A100
- S4 search: 10 days on multiple A100 GPUs
- MD simulations: Dutch supercomputer Snellius; 1.2-1.6 microseconds per ligand (Umbrella Sampling)
Artifacts
| Artifact | Type | License | Notes |
|---|---|---|---|
| S4 for de novo drug design | Code | MIT | Official PyTorch implementation with data and trained models |
| Zenodo archive | Dataset | CC-BY-4.0 | Source data and molecule designs |
Paper Information
Citation: Ozcelik, R., de Ruiter, S., Criscuolo, E., & Grisoni, F. (2024). Chemical language modeling with structured state space sequence models. Nature Communications, 15, 6176.
@article{ozcelik2024chemical,
title={Chemical language modeling with structured state space sequence models},
author={\"O{}z\c{c}elik, R{\i}za and de Ruiter, Sarah and Criscuolo, Emanuele and Grisoni, Francesca},
journal={Nature Communications},
volume={15},
number={1},
pages={6176},
year={2024},
publisher={Nature Publishing Group},
doi={10.1038/s41467-024-50469-9}
}