An Early Method for LSTM-Based Molecular Generation
This is a Method paper that applies character-level LSTM networks to the task of de novo drug-like molecule generation. The primary contribution is demonstrating that an LSTM trained on SMILES strings from a large bioactive compound database (ChEMBL) can produce novel, diverse molecules whose chemical properties closely match those of known drug-like compounds. The paper also validates the generated molecules through virtual screening with profile QSAR models, showing comparable predicted bioactivity to the training set.
The Challenge of Exploring Drug-Like Chemical Space
The theoretical space of drug-like molecules is astronomically large. Brute-force enumeration approaches such as GDB-17 (which catalogued 166 billion molecules) are feasible only for small molecules, and full enumeration of molecules with 25-30 heavy atoms (the typical size of drug molecules) remains computationally intractable. Traditional cheminformatics approaches to sampling this space rely on fragment combination, evolutionary algorithms, or particle swarm optimization.
The authors position LSTM networks as a viable alternative. LSTMs had already demonstrated the ability to learn sequential structure in domains like text and music generation, making them natural candidates for learning SMILES grammar and generating novel valid molecular strings. At the time of writing (late 2017), several groups were exploring this direction, including Bjerrum and Threlfall (ZINC-based generation), Gomez-Bombarelli et al. (VAE-based latent space design), Olivecrona et al. (RL-guided generation), and Segler et al. (focused library design). This paper contributes a large-scale empirical study with detailed analysis of the generated molecules’ chemical quality.
Character-Level LSTM with Temperature-Based Sampling
The core approach is straightforward: train an LSTM to predict the next character in a SMILES string, then sample from the trained model to generate new molecules character by character.
The network architecture consists of:
- Two stacked LSTM layers (which learn the SMILES grammar)
- A dropout layer for regularization
- A dense output layer with 23 neurons (one per character in the reduced SMILES alphabet) and softmax activation
The RMSProp optimizer was used for training. The learning rate was gradually decreased from 0.01 to 0.0002 during training. At generation time, a temperature parameter controls the randomness of character sampling to produce more diverse structures rather than reproducing training molecules too closely.
A key preprocessing step reduces the SMILES alphabet to 23 characters. Multi-character atom tokens are replaced with single characters (Cl → L, Br → R, [nH] → A). Only the organic atom subset (H, C, N, O, S, P, F, Cl, Br, I) is retained. Charged molecules, stereo information, and molecules with more than 5 ring closures are excluded. The training corpus totals 23,664,668 characters, with 40-character windows used as input sequences during training.
Training on ChEMBL and Generating One Million Molecules
Training Data
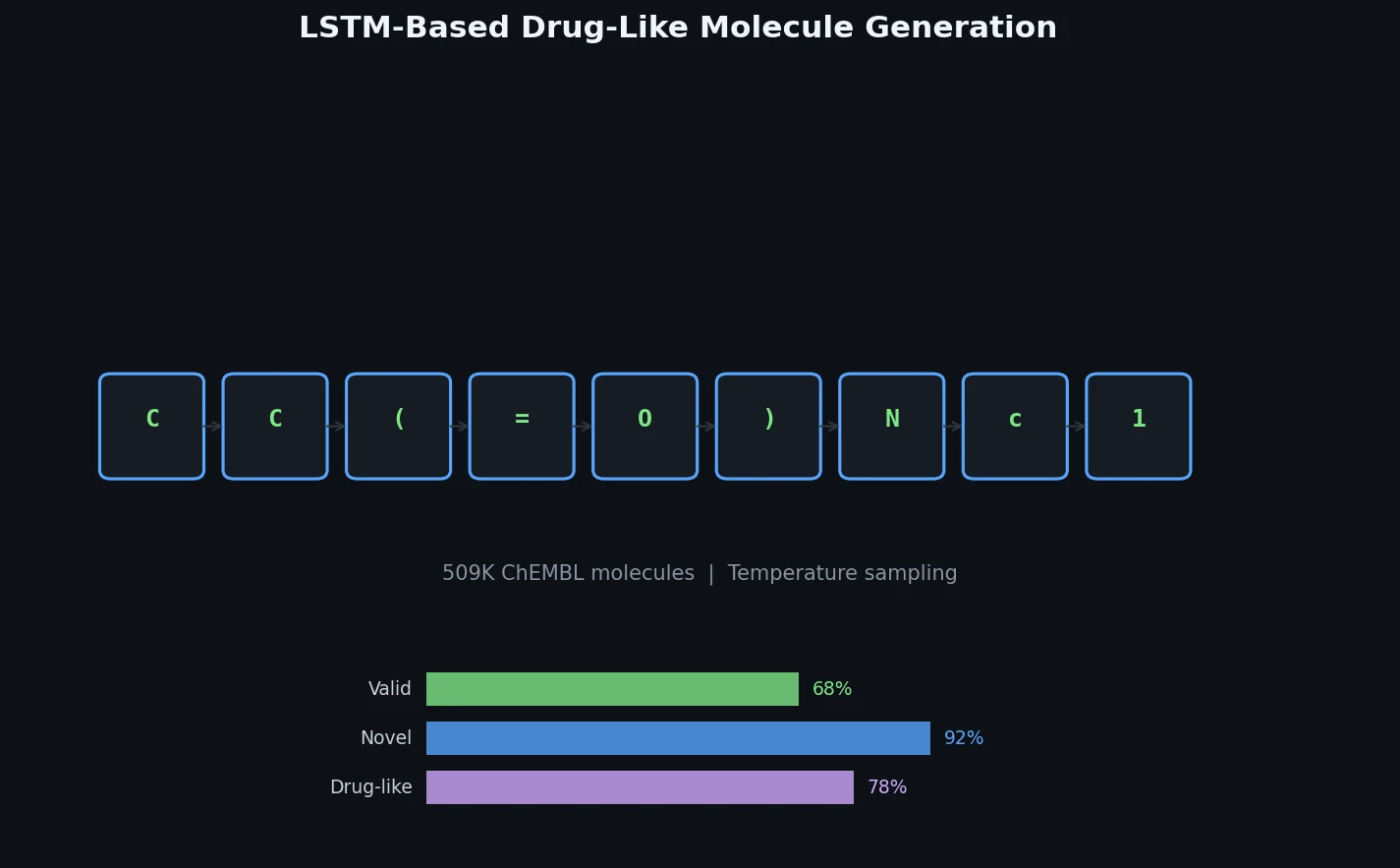
The training set consists of 509,000 bioactive molecules from ChEMBL with reported activity below 10 micromolar on any target.
Generation and Filtering
The LSTM generates SMILES strings character by character. The generated strings undergo a two-stage validation:
- Bracket and ring closure check (fast text-based): 54% of generated SMILES are discarded for unpaired brackets or ring closures
- Full chemical parsing with RDKit: An additional 14% fail due to unrealistic aromatic systems or incorrect valences
- Final yield: 32% of generated SMILES correspond to valid molecules
One million valid molecules were generated in under 2 hours on 300 CPUs.
Novelty and Diversity
Out of one million generated molecules, only 2,774 (0.28%) were identical to molecules in the training ChEMBL set. The generated set contained 627,000 unique scaffolds compared to 172,000 in ChEMBL, with an overlap of only 18,000 scaffolds. This demonstrates substantial novelty and diversity.
Physicochemical Properties
Calculated molecular descriptors (molecular weight, logP, and topological polar surface area) for the generated molecules closely matched the distributions of the ChEMBL training set. The synthetic accessibility score distributions were also practically identical, indicating comparable molecular complexity.
Substructure Feature Comparison
The paper compares substructure features across three molecule sets: ChEMBL training data, LSTM-generated molecules, and a naive SMILES baseline generator. The naive generator uses only character frequency statistics and basic SMILES syntax rules, producing primarily macrocycles with very few fused aromatic systems.
| Feature | ChEMBL (%) | LSTM Generated (%) | Naive Baseline (%) |
|---|---|---|---|
| No rings | 0.4 | 0.4 | 0.1 |
| 1 ring | 2.8 | 4.3 | 13.2 |
| 2 rings | 14.8 | 23.1 | 17.7 |
| 3 rings | 32.2 | 43.5 | 27.3 |
| 4 rings | 32.7 | 23.9 | 25.2 |
| >4 rings | 17.2 | 4.8 | 16.5 |
| Fused aromatic rings | 38.8 | 30.9 | 0.2 |
| Large rings (>8) | 0.4 | 1.8 | 75.9 |
| Spiro rings | 1.9 | 0.6 | 0.6 |
| Contains N | 96.5 | 96.1 | 92.3 |
| Contains O | 93.0 | 92.0 | 85.5 |
| Contains S | 35.6 | 27.9 | 39.6 |
| Contains halogen | 40.7 | 38.8 | 49.4 |
The LSTM-generated molecules closely mirror the ChEMBL distributions, while the naive generator fails to capture drug-like structural patterns. The LSTM tends to slightly over-represent 2-3 ring systems and under-represent 4+ ring systems relative to ChEMBL. Functional group distributions also closely matched between ChEMBL and the LSTM output.
Virtual Screening Validation
The generated molecules were evaluated using profile QSAR models for 159 ChEMBL kinase assays. The six best models (with realistic test set R-squared > 0.75) were used to predict pIC50 values for both actual ChEMBL compounds and generated compounds. The cumulative frequency distributions of predicted activity were nearly identical between the two sets.
Kolmogorov-Smirnov (KS) tests on random samples of 1,000 compounds confirmed this quantitatively:
| Assay | KS D | Distributions Differ? | Mean (Real) | Mean (Gen) | Stdev (Real) | Stdev (Gen) |
|---|---|---|---|---|---|---|
| 688395 | 6.01% | No | 4.66 | 4.69 | 0.25 | 0.24 |
| 668624 | 3.60% | No | 4.86 | 4.86 | 0.25 | 0.24 |
| 809226 | 9.90% | Yes | 5.33 | 5.26 | 0.34 | 0.30 |
| 809226 | 4.30% | No | 5.18 | 5.13 | 0.47 | 0.43 |
| 688781 | 2.20% | No | 4.83 | 4.82 | 0.26 | 0.25 |
| 809170 | 8.70% | Yes | 5.12 | 5.07 | 0.51 | 0.46 |
For 4 of 6 models, the null hypothesis that the distributions are the same could not be rejected at the 95% confidence level (critical D = 6.04%). Even for the two assays where the KS test rejected the null hypothesis, the maximum vertical distance between distributions was below 10%.
Generated Molecules Are Novel, Drug-Like, and Potentially Bioactive
The key findings of this study are:
- High novelty: Only 0.28% of generated molecules match training compounds; 627K novel scaffolds were produced versus 172K in ChEMBL
- Drug-like quality: Physicochemical properties, substructure features, functional group distributions, and synthetic accessibility scores all closely match the ChEMBL training distribution, without these being explicit constraints
- Predicted bioactivity: Virtual screening with profile QSAR models shows the generated molecules have comparable predicted activity profiles to known bioactive compounds
- Scalability: One million valid molecules in under 2 hours on 300 CPUs, with the potential to scale to billions with GPU acceleration
- LSTM superiority over naive baselines: A simple statistical SMILES generator using only character frequencies produces chemically unrealistic molecules (mostly macrocycles), demonstrating that the LSTM genuinely learns drug-like chemical patterns
The main limitations are the 32% validity rate (68% of generated SMILES are invalid), the exclusion of stereochemistry and charged molecules from the training set, and the lack of any goal-directed generation capability (the model produces unconditional samples from the training distribution). The code was described as “available on request” from the corresponding author rather than publicly released.
Reproducibility Details
Data
| Purpose | Dataset | Size | Notes |
|---|---|---|---|
| Training | ChEMBL bioactive molecules | 509,000 molecules | Activity < 10 uM on any target; organic atoms only; no charges or stereo |
Algorithms
- Double-stacked LSTM layers with dropout
- Softmax output over 23-character reduced SMILES alphabet
- RMSProp optimizer with learning rate annealed from 0.01 to 0.0002
- Temperature-based sampling at generation time
- 40-character input windows during training
Models
The architecture consists of two LSTM layers, a dropout layer, and a 23-neuron dense output layer. Exact hidden unit counts and dropout rates are not specified in the paper.
Evaluation
| Metric | Value | Notes |
|---|---|---|
| Valid SMILES rate | 32% | After bracket check and RDKit parsing |
| Novelty (vs. training) | 99.72% | Only 2,774 of 1M match ChEMBL |
| Unique scaffolds | 627,000 | vs. 172,000 in ChEMBL |
| KS test (4/6 assays) | Not significantly different | At 95% confidence |
Hardware
- Generation: 300 CPUs for under 2 hours (1 million valid molecules)
- Training hardware not specified
Paper Information
Citation: Ertl, P., Lewis, R., Martin, E., & Polyakov, V. (2017). In silico generation of novel, drug-like chemical matter using the LSTM neural network. arXiv preprint, arXiv:1712.07449.
@article{ertl2017silico,
title={In silico generation of novel, drug-like chemical matter using the LSTM neural network},
author={Ertl, Peter and Lewis, Richard and Martin, Eric and Polyakov, Valery},
journal={arXiv preprint arXiv:1712.07449},
year={2017}
}