Grammatical Evolution for De Novo Molecular Design
This is a Method paper that introduces ChemGE, a population-based molecular generation approach built on grammatical evolution. Rather than using deep neural networks, ChemGE evolves populations of SMILES strings through a context-free grammar, enabling concurrent evaluation by multiple molecular simulators and producing diverse molecular libraries. The method represents an alternative paradigm for de novo drug design: evolutionary optimization over formal grammars rather than learned latent spaces or autoregressive neural models.
Limitations of Sequential Deep Learning Generators
At the time of publication, the dominant approaches to de novo molecular generation included Bayesian optimization over VAE latent spaces (CVAE, GVAE), reinforcement learning with recurrent neural networks (ORGAN, REINVENT), sequential Monte Carlo search, and Monte Carlo tree search (ChemTS). These methods share two practical limitations:
Simulation concurrency: Most methods generate one molecule at a time, making it difficult to run multiple molecular simulations (e.g., docking) in parallel. This wastes computational resources in high-throughput virtual screening settings.
Molecular diversity: Deep learning generators tend to exploit narrow regions of chemical space. Deep reinforcement learning methods in particular often generate very similar molecules, requiring special countermeasures to maintain diversity. Since drug discovery is a multi-stage pipeline, limited diversity reduces survival rates in downstream ADMET screening.
ChemGE addresses both problems by maintaining a large population of molecules that are evolved and evaluated concurrently.
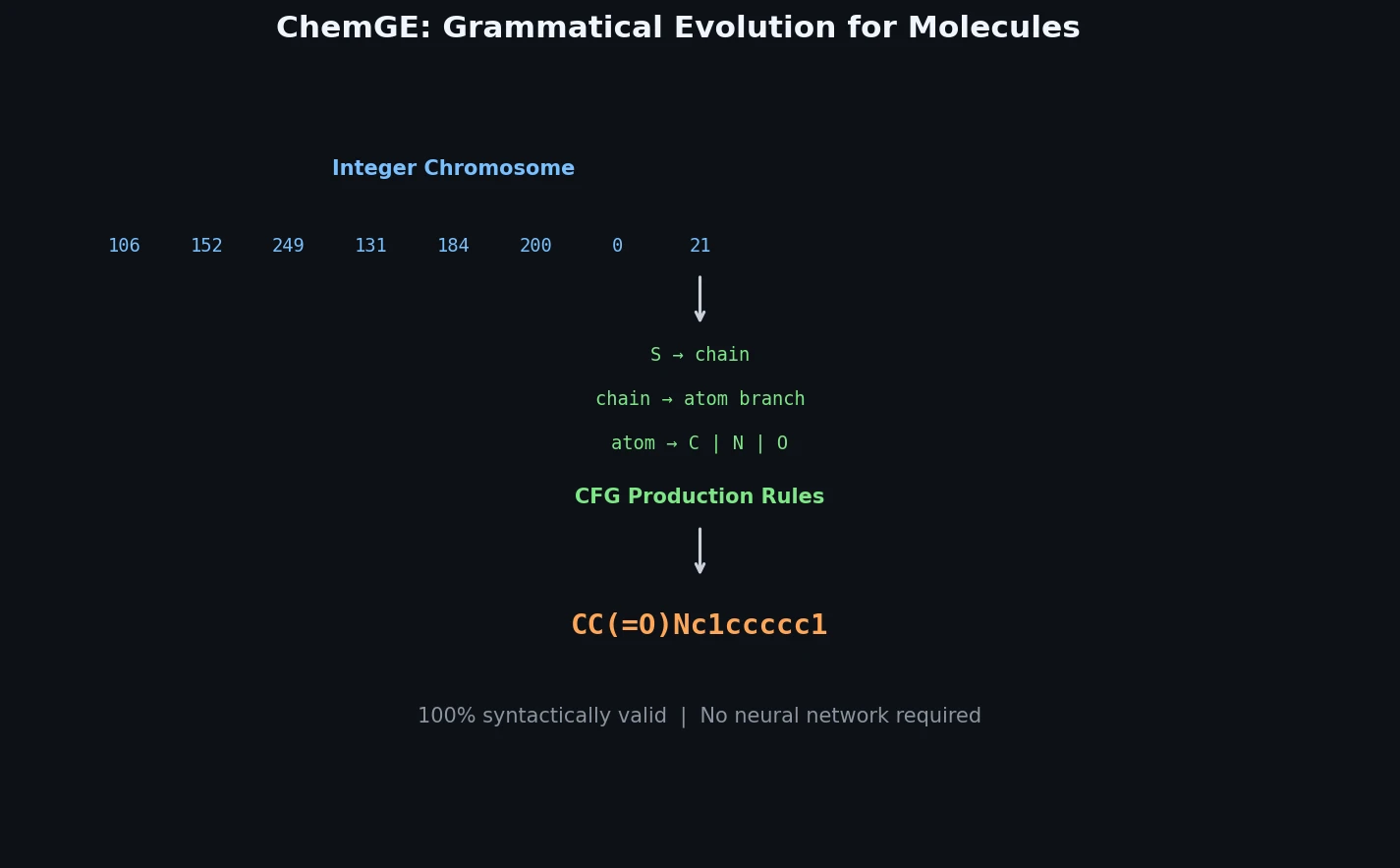
Core Innovation: Chromosome-to-SMILES Mapping via Grammar Rules
ChemGE encodes each molecule as a chromosome: a sequence of $N$ integers that deterministically maps to a SMILES string through a context-free grammar. The mapping process works as follows:
- Start with the grammar’s start symbol
- At each step $k$, look up the $k$-th integer $c = C[k]$ from the chromosome
- Identify the leftmost non-terminal symbol and count its $r$ applicable production rules
- Apply the $((c \bmod r) + 1)$-th rule
- Repeat until no non-terminal symbols remain or the chromosome is exhausted
The context-free grammar is a subset of the OpenSMILES specification, defined formally as $G = (V, \Sigma, R, S)$ where $V$ is the set of non-terminal symbols, $\Sigma$ is the set of terminal symbols, $R$ is the set of production rules, and $S$ is the start symbol.
Evolution follows the $(\mu + \lambda)$ evolution strategy:
- Create $\lambda$ new chromosomes by drawing random chromosomes from the population and mutating one integer at a random position
- Translate each chromosome to a SMILES string and evaluate fitness (e.g., docking score). Invalid molecules receive fitness $-\infty$
- Select the top $\mu$ molecules from the merged pool of $\mu + \lambda$ candidates
The authors did not use crossover, as it did not improve performance. Diversity is inherently maintained because a large fraction of molecules are mutated in each generation.
Experimental Setup and Benchmark Comparisons
Druglikeness Score Benchmark
The first experiment optimized the penalized logP score $J^{\log P}$, an indicator of druglikeness defined as:
$$ J^{\log P}(m) = \log P(m) - \text{SA}(m) - \text{ring-penalty}(m) $$
where $\log P(m)$ is the octanol-water partition coefficient, $\text{SA}(m)$ is the synthetic accessibility score, and ring-penalty$(m)$ penalizes carbon rings larger than size 6. All terms are normalized to zero mean and unit standard deviation. Initial populations were randomly sampled from the ZINC database (35 million compounds), with fitness set to $-\infty$ for molecules with molecular weight above 500 or duplicate structures.
ChemGE was compared against CVAE, GVAE, and ChemTS across population sizes $(\mu, \lambda) \in {(10, 20), (100, 200), (1000, 2000), (10000, 20000)}$.
| Method | 2h | 4h | 6h | 8h | Mol/Min |
|---|---|---|---|---|---|
| ChemGE (10, 20) | 4.46 +/- 0.34 | 4.46 +/- 0.34 | 4.46 +/- 0.34 | 4.46 +/- 0.34 | 14.5 |
| ChemGE (100, 200) | 5.17 +/- 0.26 | 5.17 +/- 0.26 | 5.17 +/- 0.26 | 5.17 +/- 0.26 | 135 |
| ChemGE (1000, 2000) | 4.45 +/- 0.24 | 5.32 +/- 0.43 | 5.73 +/- 0.33 | 5.88 +/- 0.34 | 527 |
| ChemGE (10000, 20000) | 4.20 +/- 0.33 | 4.28 +/- 0.28 | 4.40 +/- 0.27 | 4.53 +/- 0.26 | 555 |
| CVAE | -30.18 +/- 26.91 | -1.39 +/- 2.24 | -0.61 +/- 1.08 | -0.006 +/- 0.92 | 0.14 |
| GVAE | -4.34 +/- 3.14 | -1.29 +/- 1.67 | -0.17 +/- 0.96 | 0.25 +/- 1.31 | 1.38 |
| ChemTS | 4.91 +/- 0.38 | 5.41 +/- 0.51 | 5.49 +/- 0.44 | 5.58 +/- 0.50 | 40.89 |
At $(\mu, \lambda) = (1000, 2000)$, ChemGE achieved the highest final score of 5.88 and generated 527 unique molecules per minute, roughly 13x faster than ChemTS and 3700x faster than CVAE. The small population (10, 20) converged prematurely with insufficient diversity, while the overly large population (10000, 20000) could not run enough generations to optimize effectively.
Docking Experiment with Thymidine Kinase
The second experiment applied ChemGE to generate molecules with high predicted binding affinity for thymidine kinase (KITH), a well-known antiviral drug target. The authors used rDock for docking simulation, taking the best intermolecular score $S_{\text{inter}}$ from three runs with different initial conformations. Fitness was defined as $-S_{\text{inter}}$ (lower scores indicate higher affinity). The protein structure was taken from PDB ID 2B8T.
With 32 parallel cores and $(\mu, \lambda) = (32, 64)$, ChemGE completed 1000 generations in approximately 26 hours, generating 9466 molecules total. Among these, 349 molecules achieved intermolecular scores better than the best known inhibitor in the DUD-E database.
Diversity Analysis
Molecular diversity was measured using internal diversity based on Morgan fingerprints:
$$ I(A) = \frac{1}{|A|^2} \sum_{(x,y) \in A \times A} T_d(x, y) $$
where $T_d(x, y) = 1 - \frac{|x \cap y|}{|x \cup y|}$ is the Tanimoto distance.
The 349 “ChemGE-active” molecules (those scoring better than the best known inhibitor) had an internal diversity of 0.55, compared to 0.46 for known inhibitors and 0.65 for the whole ZINC database. This is a substantial improvement over known actives, achieved without any explicit diversity-promoting mechanism.
ISOMAP visualizations showed that ChemGE populations migrated away from known inhibitors over generations, ultimately occupying a completely different region of chemical space by generation 1000. This suggests ChemGE discovered a novel structural class of potential binders.
High Throughput and Diversity Without Deep Learning
ChemGE demonstrates several notable findings:
Deep learning is not required for competitive de novo molecular generation. Grammatical evolution over SMILES achieves higher throughput and comparable or better optimization scores than VAE- and RNN-based methods.
Population size matters significantly. Too small a population leads to premature convergence. Too large a population prevents sufficient per-molecule optimization within the computational budget. The $(\mu, \lambda) = (1000, 2000)$ setting provided the best balance.
Inherent diversity is a key advantage of evolutionary methods. Without any explicit diversity loss or penalty, ChemGE maintains diversity comparable to the ZINC database and exceeds that of known active molecules.
Parallel evaluation is naturally supported. Each generation produces $\lambda$ independent molecules that can be evaluated by separate docking simulators simultaneously.
The authors acknowledge several limitations. Synthetic routes and ADMET properties were not evaluated for the generated molecules. The docking scores, while favorable, require confirmation through biological assays. The authors also note that incorporating probabilistic or neural models into the evolutionary process might further improve performance.
Reproducibility Details
Data
| Purpose | Dataset | Size | Notes |
|---|---|---|---|
| Initial population | ZINC | ~35M compounds | Randomly sampled starting molecules |
| Docking target | PDB 2B8T | 1 structure | Thymidine kinase-ligand complex |
| Baseline actives | DUD-E (KITH) | 57 inhibitors | Known thymidine kinase inhibitors |
Algorithms
- Grammatical evolution with $(\mu + \lambda)$ evolution strategy
- Mutation only (no crossover)
- Context-free grammar subset of OpenSMILES specification
- Chromosome length: $N$ integers per molecule
- Fitness set to $-\infty$ for invalid SMILES, MW > 500, or duplicate molecules
Models
No neural network models are used. ChemGE is purely evolutionary.
Evaluation
| Metric | Value | Baseline | Notes |
|---|---|---|---|
| Max $J^{\log P}$ (8h) | 5.88 +/- 0.34 | ChemTS: 5.58 +/- 0.50 | ChemGE (1000, 2000) |
| Molecules/min | 527 | ChemTS: 40.89 | ~13x throughput improvement |
| Docking hits | 349 | Best DUD-E inhibitor | Molecules with better $S_{\text{inter}}$ |
| Internal diversity | 0.55 | Known inhibitors: 0.46 | Morgan fingerprint Tanimoto distance |
Hardware
- CPU: Intel Xeon E5-2630 v3 (benchmark experiments, single core)
- Docking: 32 cores in parallel (thymidine kinase experiment, ~26 hours for 1000 generations)
Artifacts
| Artifact | Type | License | Notes |
|---|---|---|---|
| ChemGE | Code | MIT | Official implementation |
Paper Information
Citation: Yoshikawa, N., Terayama, K., Sumita, M., Homma, T., Oono, K., & Tsuda, K. (2018). Population-based de novo molecule generation, using grammatical evolution. Chemistry Letters, 47(11), 1431-1434. https://doi.org/10.1246/cl.180665
@article{yoshikawa2018chemge,
title={Population-based De Novo Molecule Generation, Using Grammatical Evolution},
author={Yoshikawa, Naruki and Terayama, Kei and Sumita, Masato and Homma, Teruki and Oono, Kenta and Tsuda, Koji},
journal={Chemistry Letters},
volume={47},
number={11},
pages={1431--1434},
year={2018},
publisher={Oxford University Press},
doi={10.1246/cl.180665}
}