Language Models as 3D Chemical Structure Generators
This is a Method paper that demonstrates transformer-based language models can generate molecules, crystalline materials, and protein binding sites directly in three dimensions by training on sequences derived from standard chemical file formats (XYZ, CIF, PDB). The key contribution is showing that unmodified autoregressive language models, using only next-token prediction, achieve performance comparable to domain-specific 3D generative models that incorporate SE(3) equivariance and other geometric inductive biases.
Beyond Graphs and Strings: The Need for 3D Chemical Generation
Molecular design with deep learning has largely relied on two representation paradigms: molecular graphs (processed with graph neural networks) and linearized string representations like SMILES and SELFIES (processed with sequence models). Both approaches have proven effective for drug-like organic molecules, but they share a fundamental limitation: they cannot represent structures whose identity depends on 3D spatial arrangement.
Crystalline materials, for example, have periodic lattice structures that cannot be reduced to simple graphs. Protein binding sites are defined by the 3D arrangement of hundreds of atoms across multiple residues. For tasks like catalysis design or structure-based drug discovery, the geometric positions of atoms are essential information that graphs and strings discard entirely.
Existing 3D generative models address this gap but typically require specialized architectures with SE(3) equivariance to handle rotational and translational symmetries. This work asks whether the general-purpose sequence modeling capability of transformers is sufficient to learn 3D chemical structure distributions without any domain-specific architectural modifications.
Direct Tokenization of Chemical File Formats
The core insight is straightforward: any 3D molecule, crystal, or biomolecule is already stored as text in standard file formats (XYZ, CIF, PDB). These files encode atom types and their Cartesian coordinates as sequences of characters and numbers. Rather than designing specialized architectures for point cloud generation, the authors simply tokenize these files and train a standard GPT-style transformer to predict the next token.
A molecule with $n$ atoms is represented as:
$$ \mathcal{M} = (e_1, x_1, y_1, z_1, \dots, e_n, x_n, y_n, z_n) $$
where $e_i$ is the element type and $(x_i, y_i, z_i)$ are Cartesian coordinates. Crystals additionally include lattice parameters:
$$ \mathcal{C} = (\ell_a, \ell_b, \ell_c, \alpha, \beta, \gamma, e_1, x_1, y_1, z_1, \dots, e_n, x_n, y_n, z_n) $$
Protein binding sites use residue-atom indicators (e.g., HIS-C, CYS-N) instead of bare element symbols:
$$ \mathcal{P} = (a_1, x_1, y_1, z_1, \dots, a_n, x_n, y_n, z_n) $$
The language model learns the joint distribution via autoregressive factorization:
$$ p(x) = \prod_{i=1}^{n} p(t_i \mid t_{i-1}, \dots, t_1) $$
Two tokenization strategies are explored:
- Character-level (LM-CH): Every character in the file is a token, including digits, minus signs, spaces, and newlines. This produces long sequences but uses a small vocabulary (~30 tokens).
- Atom+coordinate-level (LM-AC): Each atom placement requires exactly 4 tokens: one element/residue token and three coordinate tokens (e.g., ‘-1.98’). The vocabulary is larger (~100-10K tokens) but sequences are shorter.
Numerical precision is controlled by rounding coordinates to 1, 2, or 3 decimal places. Since the model lacks rotation and translation invariance, random rotation augmentation during training improves performance.
Experiments Across Molecules, Crystals, and Protein Binding Sites
Molecular Generation (ZINC)
The model is evaluated on 250K commercially available molecules from the ZINC dataset, with an average of 23 heavy atoms. XYZ files are generated using RDKit’s conformer tools. Coordinates use 2 decimal places of precision. The authors generate 10K molecules and evaluate both 3D geometry quality and standard generative metrics.
For 3D geometry assessment, root mean squared deviation (RMSD) between language model-generated conformers and RDKit-generated conformers shows most molecules fall between 1.0 and 2.0 RMSD, with a heavy tail extending to 4.0.
Standard metrics include validity, uniqueness, novelty, and earth mover’s distance (WA) for molecular property distributions (QED, SA score, molecular weight).
| Model | 3D | Valid (%) | Unique (%) | Novel (%) | WA MW | WA SA | WA QED |
|---|---|---|---|---|---|---|---|
| Train | No | 100.0 | 100.0 | 100.0 | 0.816 | 0.013 | 0.002 |
| SM-LM | No | 98.35 | 100.0 | 100.0 | 3.640 | 0.049 | 0.005 |
| SF-LM | No | 100.0 | 100.0 | 100.0 | 3.772 | 0.085 | 0.006 |
| JTVAE | No | 100.0 | 98.56 | 100.0 | 22.63 | 0.126 | 0.023 |
| ENF | Yes | 1.05 | 96.37 | 99.72 | 168.5 | 1.886 | 0.160 |
| G-SchNet | Yes | 1.20 | 55.96 | 98.33 | 152.7 | 1.126 | 0.185 |
| EDM | Yes | 77.51 | 96.40 | 95.30 | 101.2 | 0.939 | 0.093 |
| LM-CH | Yes | 90.13 | 100.0 | 100.0 | 3.912 | 2.608 | 0.077 |
| LM-AC | Yes | 98.51 | 100.0 | 100.0 | 1.811 | 0.026 | 0.004 |
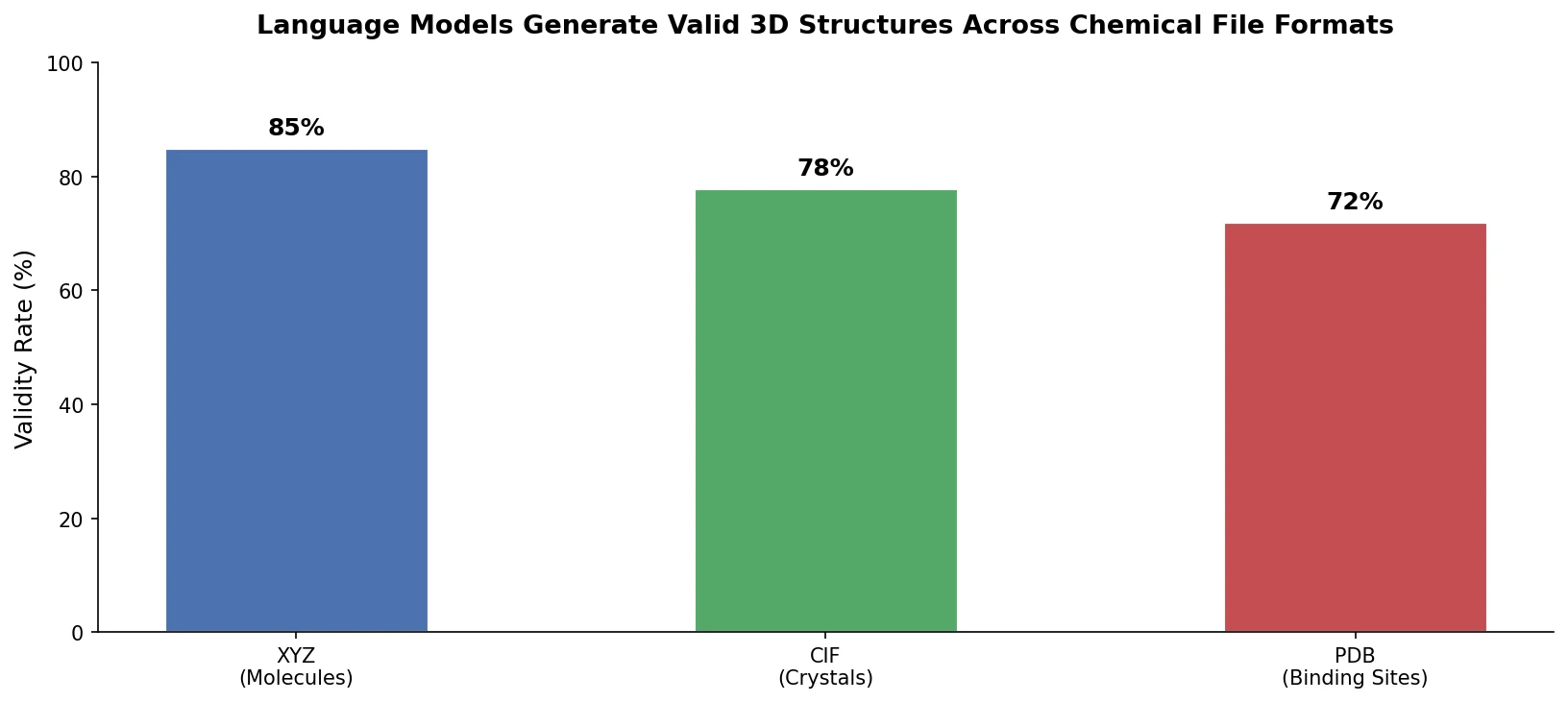
The atom+coordinate tokenization model (LM-AC) achieves 98.51% validity with 100% uniqueness and novelty. Its WA scores for molecular weight (1.811) and QED (0.004) are substantially better than all other 3D generative baselines and competitive with SMILES/SELFIES language models. The character-level model (LM-CH) at 90.13% validity performs comparably to graph-based models but falls short of the string-based language models.
Crystal Generation (Perov-5 and MP-20)
Crystal generation uses CIF-derived sequences with 3 decimal places of precision. Two datasets are used: Perov-5 (18,928 perovskite materials, 5 atoms per unit cell, 56 elements) and MP-20 (45,231 diverse materials, 1-20 atoms per unit cell, 89 elements).
Evaluation metrics include structural validity (minimum interatomic distance > 0.5 angstrom), compositional validity (charge neutrality via SMACT), coverage (recall and precision between generated and test sets), and earth mover’s distance for density and number of unique elements.
| Data | Model | Struc. Valid (%) | Comp. Valid (%) | COV-R (%) | COV-P (%) | WA density | WA elements |
|---|---|---|---|---|---|---|---|
| Perov-5 | CDVAE | 100.0 | 98.59 | 99.45 | 98.46 | 0.126 | 0.063 |
| Perov-5 | LM-CH | 100.0 | 98.51 | 99.60 | 99.42 | 0.071 | 0.036 |
| Perov-5 | LM-AC | 100.0 | 98.79 | 98.78 | 99.36 | 0.089 | 0.028 |
| MP-20 | CDVAE | 100.0 | 86.70 | 99.15 | 99.49 | 0.688 | 1.432 |
| MP-20 | LM-CH | 84.81 | 83.55 | 99.25 | 97.89 | 0.864 | 0.132 |
| MP-20 | LM-AC | 95.81 | 88.87 | 99.60 | 98.55 | 0.696 | 0.092 |
On Perov-5, both language models outperform CDVAE across most metrics. On the more diverse MP-20 dataset, LM-AC achieves the best scores on 3 of 6 metrics and remains competitive on the others. LM-CH struggles more with structural validity on MP-20 (84.81%).
Protein Binding Site Generation (PDB)
The most challenging task involves generating protein binding sites (~200-250 atoms each) from PDB-derived sequences. The dataset contains approximately 180K protein-ligand pairs. Residue-atom tokenization is used (e.g., CYS-C, CYS-N), with 2 decimal places of precision.
Validity is assessed per-residue using xyz2mol, with an additional check for inter-residue atomic overlap (atoms from different residues closer than the minimum bond distance). Approximately 99% of generated pockets pass the residue validity check, while about 5% fail the overlap check. Of generated pockets, 89.8% have unique residue orderings, and 83.6% have novel orderings not seen in training, indicating the model is generating novel binding site structures rather than memorizing.
Competitive 3D Generation Without Geometric Inductive Biases
The central finding is that standard transformer language models, without any equivariance or geometric inductive biases, can generate valid 3D chemical structures across three substantially different domains. The atom+coordinate tokenization (LM-AC) consistently outperforms character-level tokenization (LM-CH), likely because it produces shorter sequences and reduces the number of sequential decisions needed per atom placement.
Several limitations are worth noting. The model generates atoms using absolute Cartesian coordinates, which means it must learn rotation and translation invariance purely from data augmentation rather than having it built into the architecture. The authors acknowledge this becomes increasingly difficult as structure size grows. The vocabulary size also scales with coordinate precision and structure complexity, which could become prohibitive for very large systems.
The paper does not include computational cost comparisons with baseline models, making it difficult to assess the practical tradeoff between the simplicity of the language modeling approach and the efficiency of specialized architectures. The authors also note that further validation through computational simulation and experiment is needed to confirm the physical plausibility of generated structures.
Future directions identified include inverse design of molecules and materials conditioned on target properties, extension to more complex structures (metal-organic frameworks), and exploration of alternative tokenization strategies to handle larger systems.
Reproducibility Details
Data
| Purpose | Dataset | Size | Notes |
|---|---|---|---|
| Training/Eval | ZINC | 250K molecules | ~23 heavy atoms avg; XYZ files via RDKit conformer generation |
| Training/Eval | Perov-5 | 18,928 perovskites | 5 atoms/unit cell, 56 elements |
| Training/Eval | MP-20 | 45,231 materials | 1-20 atoms/unit cell, 89 elements |
| Training/Eval | Protein binding sites | ~180K protein-ligand pairs | Processed to 200-250 atoms per pocket |
Algorithms
- Architecture: GPT-style transformer with ~1M to 100M parameters
- Layers: 12
- Embedding size: 128 to 1024
- Attention heads: 4 to 12
- Batch size: 4 to 32 structures
- Learning rate: $10^{-4}$ to $10^{-5}$, decayed to $9 \times 10^{-6}$
- Data augmentation: Random rotation of training structures at each epoch
- Numerical precision: 2 decimal places (molecules, proteins), 3 decimal places (crystals)
Models
No pre-trained model weights are publicly available. The paper mentions “Example code can be found at” but the URL appears to be missing from the published version.
Evaluation
| Metric | Domain | Description |
|---|---|---|
| Validity | Molecules | xyz2mol produces valid RDKit Mol object |
| Validity | Crystals | Structural (min distance > 0.5 angstrom) and compositional (charge neutral) |
| Uniqueness | All | Fraction of distinct generated structures |
| Novelty | All | Fraction not in training set |
| Earth mover’s distance | All | Distribution match for domain-specific properties |
| RMSD | Molecules | Deviation from RDKit conformer geometries |
| Coverage | Crystals | Recall and precision between generated and test sets |
Hardware
Models were trained using the Canada Computing Systems (Compute Canada). Specific GPU types, counts, and training times are not reported.
Artifacts
No public code repository, model weights, or datasets specific to this work were found. The ZINC, Perov-5, and MP-20 datasets used for evaluation are publicly available from their original sources.
Paper Information
Citation: Flam-Shepherd, D. & Aspuru-Guzik, A. (2023). Language models can generate molecules, materials, and protein binding sites directly in three dimensions as XYZ, CIF, and PDB files. arXiv preprint arXiv:2305.05708.
@article{flamshepherd2023language,
title={Language models can generate molecules, materials, and protein binding sites directly in three dimensions as {XYZ}, {CIF}, and {PDB} files},
author={Flam-Shepherd, Daniel and Aspuru-Guzik, Al{\'a}n},
journal={arXiv preprint arXiv:2305.05708},
year={2023}
}